Acute kidney injury (AKI) is a widely held concern related to a substantial burden of morbidity, mortality and expenditure in the healthcare system. AKI is not a simple illness but a complex conglomeration of syndromes that often occurs as part of other syndromes in its wide clinical spectrum of the disease. Genetic factors have been suggested as potentially responsible for its susceptibility and severity.
- acute kidney injury
- predisposition
- gene polymorphism
- genetic variation
- intensive care unit
1. Introduction
Disparities in kidney disease predisposition among individuals have been sharply identified [1]. The United States Renal Data System Report shows a constant increment in the occurrence of end-stage kidney disease (ESKD) among Africans that is 3.5–5-fold that of Europeans [2]. While the risk of Africans developing ESKD is 8% approximately, that of Europeans is nearly 2–3% [3].
Acute kidney injury (AKI) is a widely held concern in hospitalized patients [4]. Nearly 40% of patients in intensive care units (ICUs) develop AKI, as well as other concomitant states such as sepsis or hemodynamic failure [5][6][15,16]. AKI is presented by a sharp and unexpected slump in renal function, often related to other medical conditions, such as septic shock, or procedures, for instance cardiac surgery [7][17]. Nevertheless, why certain patients are more vulnerable to develop AKI than others remains unknown [8][20].
Despite progress in the comprehension of AKI pathophysiology, its pathogenesis remains complex and incompletely understood [9][10][11][21,22,23], including numerous pathophysiological mechanisms that lead to sudden tubular necrosis/apoptosis and kidney dysfunction [8][12][13][14][20,24,25,26]. These comprise ischemia/reperfusion injury, complement activation, adenosine triphosphate (ATP) reduction, infiltration of leukocytes, generation of endotoxins, oxygen radicals, proinflammatory mediator generation, endothelial damage, fibrosis, microcirculatory dysfunction and, finally, cell death [12][15][16][17][24,27,28,29].
Recently, inflammatory response has been recognized as a crucial factor in AKI pathogenesis [13][18][19][20][21][22][25,30,31,32,33,34]. Infiltration of inflammatory cells has been identified in the injured kidney, which causes renal vessel damage and posterior progress to AKI [23][35]. These cells do not only start but also maintain the kidney damage by producing oxygen radical species, vasoconstrictors such as endothelin and impeding the release of nitric oxide, directly causing endothelial injury [18][30]. What is more, patients’ individual host repair and regeneration biology could also have a major influence on the etiology of AKI [24][36].
Genetic factors have been proposed as potentially responsible for the susceptibility and severity of AKI, explaining why only particular patients are more prone to AKI and why different patients respond to treatment distinctly [25][26][27][28][29][37,38,39,40,41]. This fact has led to a new form of exercise medicine, known as personalized medicine, the aim of which is for patients to be treated as individuals and not as groups [30][42]. A deeper understanding of how gene interaction drives health or disease might lead to a more personalized medical strategy, allowing the creation of a unique individual genetic print on which medical decisions should be based. Numerous articles have explored the relationship between different polymorphisms and AKI predisposition in various clinical scenarios
Polymorphism association studies usually consist of case–control and observational studies, comparing the occurrence of a genetic variation in some individuals with a certain illness to the occurrence in a control healthy population. Furthermore, although several polymorphisms have been identified, most studies often consist of relatively small homogeneous sample size populations, which significantly restricts the ability to draw conclusions from the results of the general population and frequently gives conflicting results with mainly nonsignificant findings. What is more, some diseases are polygenic in nature, and the interactions among relevant proposed genes have not been identified. Indeed, a far more complete overview could be the application of linking analyses, in which genetically related polymorphisms are studied and where the polymorphism of a particular gene could be the promoter of another illness-inducing variation [31][45].
Genome-wide association study (GWAS) has come to revolutionize the identification of novel disease susceptibility genes, leading to the discovery of novel biological mechanisms and offering insight into the ethnic variation of complex traits, among others. In spite of the fact that GWAS is a powerful method to determine genotype–phenotype association, this method presents limitations such as that it can only explain a small fraction of the heritability of complex traits and does not necessarily pinpoint causal variants and genes. Moreover, it was necessary to adopt a high level of significance to account for multiple tests. This is because false-positive relationships are innumerable in genetic association reports to identify frequent polymorphisms [32][46].
Association of genetic polymorphisms and AKI risk usually differs among ethnicities, populations and geographical boundaries [16][33][34][28,50,51]. This is of paramount importance, as some genetic variations are uncommon in certain racial groups but not in others. Most article clinical settings refer to cardiac surgery patients, critically ill patients and contrast-induced AKI, which is the third most common cause of AKI.
Chang et al. found that patients with contrast-induced AKI presented a higher decline in kidney function than those without contrast media [35][52]. What is more, Wu and coworkers confirmed in 2018 that baseline serum creatinine is an independent risk factor for contrast-induced AKI [19][31]. Men with baseline creatinine ≥114.9 µmol/L and women with creatinine
Several studies have attempted to quantify the contribution of genetic and environmental risk factors to diseases [36][37][38][54,55,56]. Likewise, environmental factors can invoke heritable phenotype changes in DNA without alterations in its sequence. In this sense, epigenetics entails profound changes that affect gene activity and expression. Therefore, environmental determinants induce epigenetic marks that can trigger the development of certain illnesses.
2. Associated Genes
While an individual’s genotype depicts the combination of parental genotypes, two distinct individuals have >99.9% of their DNA sequences [39][59]. Variants observed in the left 0.1% of the human genome are known as gene polymorphisms and have become the topic of intense investigation. Indeed, such variations are markers of biological variety, and some genotypic polymorphisms have been identified to be related to particular human disease phenotypes [16][28]. It is unclear if any of these genetic polymorphisms are involved in the etiology of certain illnesses, as they may be placed close to other pathogenic genetic factors, known as linkage disequilibrium [16][28].
Variants can appear at one or more of the following locations: (1) the promoter region, (2) the exon(s) or the gene coding region, (3) the intron(s) or the gene intervening sequences and (4) the 3′-untranslated (3′-UTR) region (Figure 1).
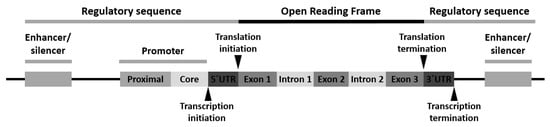
Polymorphism of the promoter region may affect the transcriptional activity. Polymorphism of the exons or encoding regions may be mute or affect gene expression or function. As introns are transcribed but removed from the messenger RNA (mRNA) before it is translated into a protein, its polymorphism may cause defects in RNA and mRNA processing. Finally, polymorphism in the 3′-UTR region may alter the RNA expectancy or influence the mRNA ribosomal translation [16][28].
Three classes of gene variants have been reported: (1) single-nucleotide polymorphism (SNP), (2) variable number of tandem repeats (VNTRs) or minisatellite polymorphism and (3) microsatellite polymorphism, with SNP being the most common [16][28].
AKI susceptibility and severity are related to multiple genetic factors that are involved in several pathophysiological mechanisms as follows.
Given the relevance of inflammatory processes in the development of AKI, polymorphisms in inflammation-related genes might influence the predisposition of an individual to AKI [8][20].
Among the important inflammation-related genes that could play a role in AKI are IL6, IL10, NFBK1, NFKBIA, IL18 and TNF.
Even rare polymorphisms with very low minor allele frequencies could provide vital information and potential usefulness as a marker in the investigation of genetic predisposition to AKI [40][60].
did not find any association between isolated or combined IL6 with other genetic polymorphisms and AKI development [41][62], Nechemia-Arbely et al. identified an association between IL6 and AKI development [42][63]. IL6-174 G/C polymorphism regulated postoperative IL6 levels and was related to the severity of postoperative AKI and length of hospital stay following coronary artery surgery [43][65]. Three promoter polymorphisms within theIL6genes, namely rs1800795, rs1800796 and rs1800797 polymorphisms, have been identified to influence the expression and secretion of the cytokine [44][66]. Found that a combination of angiotensinogen (AGT) gene +842C allele (rs699) and IL-6–572C allele in Caucasians is related to kidney impairment.
The IL-10 gene is located on chromosome 1q31-32 [45][68], and the variation in IL-10 production is genetically set up and controlled at the transcriptional region [16][28]. The IL-10 promoter site is polymorphic with a single-base-pair replacement at position –1082 (G to A).
Interleukin is implicated in AKI pathogenesis due to its anti-inflammatory role, as interleukin-10 facilitates the inhibition of immune cells and secretion of proinflammatory mediators, interrupting the healing process after kidney injury [46][70]. Promoter polymorphisms within theIL10gene, namely rs1800896 and rs3021097 polymorphisms, have been demonstrated to influence the level of the interleukin [8][20]. Similarly, Hashad and colleagues have demonstrated that the low-producer genotype of IL-10 (–1082 G/A) variants was a predisposing factor for AKI in ICU patients with severe sepsis.
The TNF-α gene is placed on the short arm of chromosome 6. Variants located in the promoter region of the TNF-α gene at positions −238 (G to A) and −308 (G to A) have been described. The −308 A allele, known as the TNF-α2 allele, increases promoter activity, boosts TNF·α production and has been related to superior serum creatinine and urinary kidney injury molecule-1 (KIM-1) levels and greater multiorgan failure calculations in patients with AKI [16][47][28,72].
TNF-α gene variants may alter variations in the proinflammatory cytokine reaction to stressful stimulation. This may have enormous implications in AKI presentation, as the intensity of proinflammatory reactions may determine the graveness of AKI and, therefore, the demand for renal replacement therapy and in-hospital mortality [16][28].Figure 2 depicts an overview of the interstitial inflammation caused by TNF-α.
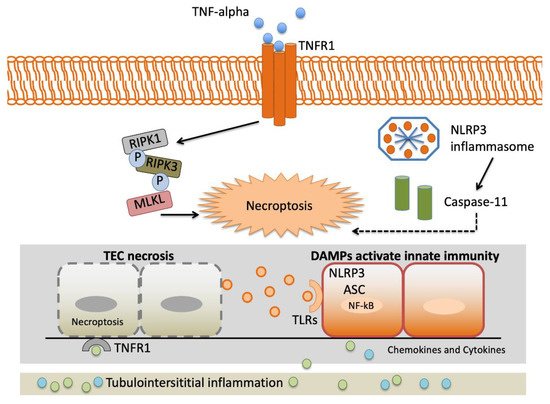
The TNF-α gene rs1800629 identified by Jaber and colleagues [16][28] is one of the most frequently studied polymorphisms in AKI [41][48][47][49][50][51][52][62,71,72,73,74,75,76]. It is related to superior levels of TNF-α in vitro, AKI predisposition and increased mortality in patients with renal replacement therapy (RRT) demonstrated rs1800629 polymorphism is a predisposing factor for AKI in ICU patients with severe sepsis [48][71].
IFN-γ is related to inflammatory response and renal damage [53][78]. Similarly, LT-α or TGF-β prompts neutrophils, T cells, monocytes and fibroblasts chemotaxis to the injury site [54][79]. Grabulosa and coworkers have shown that, although higher frequencies of polymorphisms of rs1800470, rs1800471 from the TGF-β and rs2430561 from IFN-γ were observed in critically ill patients, none was significantly associated as a risk factor for AKI [55][80].
It serves as a ligand for the T-cell receptor (TCR) and is involved in multiple autoimmune conditions. In addition, HLA-DR expression is an integral part of the glomerular capillary and peritubular endothelium [56][82]. In acute inflammation states, such as AKI, HLA-DR expression is exerted. Nevertheless, HLA-DRB alleles were found to be associated with less requirement of RRT [57][83].
NFKB1 encodes for nuclear factor kappa beta 1 Although it does not play a direct role in inflammation, it serves as the central regulator of a huge number of molecules involved in the inflammatory process. NF-κB1 functions as a central regulator for the activation and coordination of a vast assembling of genes involved in pro- and anti-inflammatory processes, including but not restricted to TNF, IL-1β and IL-6 [58][84]. NF-κB1 participates in the inflammation process via various signaling pathways; therefore, its related genes are intimately connected with the AKI pathogenesis [59][60][61][62][85,86,87,88].
The cellular level of NF-κB1 (also named p50 protein) is tightly controlled by IκBα, which is encoded by NFKBIA [63][89]. The rs2233406 and rs696 polymorphisms of the NF-BIA gene are, respectively, placed at the promoter and 3′UTR region of the gene.
NFKB1rs28362491,NFKBIArs2233406 andNFKBIArs696 polymorphisms were related to reduced predisposition of AKI among Chinese children [8][20]. The NFKB1 rs28362491 This insertion decreases the binding affinity of the promoter sequence and leads to a reduced NFKB1 promoter activity, which results in a low inflammation activity. This exemplifies the complexity of the interactions by which genetic polymorphisms could affect disease susceptibility.
Macrophage migration inhibitory factor (MIF) is a cytokine implicated in various inflammatory processes that is rapidly released from preformed intracellular pools in response to multiple cellular and systemic noxious stimuli, including ischemia/reperfusion, endotoxemia and surgery. For instance, cardiac surgery generates an increase in MIF serum levels [64][90].
Averdunk et al. showed that macrophage migration inhibitory factor (MIF) promoter polymorphisms (rs3063368, rs755622) are associated with AKI and death after cardiac surgery [65][91]. MIF may mediate AKI via CD74/TLR4-NF-KB pathway [66][92].
Interleukin-18, encoded byIL18, is also implicated in AKI pathogenesis. Studies have shown that interleukin-18 is linked to AKI, inducing kidney acute tubular necrosis [67][68][93,94]. Thus, a disrupted level of interleukin-18 could serve as a risk factor for AKI. Two suchIL18polymorphisms are the rs1946518 and rs187238 polymorphisms.
Vascular endothelial growth factor (VEGF) is a protein that promotes angiogenesis, vessel permeability, cellular survival and differentiation [69][70][95,96]. The rs3025039 genotype has been shown to boost AKI predisposition in critically ill patients with severe sepsis [49][73].
The AGT gene is located on chromosome 1 band q42. It encodes the angiotensinogen precursor, which is a fundamental component of the renin–angiotensin–aldosterone system (RAAS), being a potent vasoconstrictor. It is a major regulator of blood pressure, fluid and electrolyte homeostasis, playing a key role in renal disease pathology (Figure 3). While AGT 842C was identified by Stafford-Smith [71][67] as a major risk factor to develop postoperative renal injury, Isbir et al.
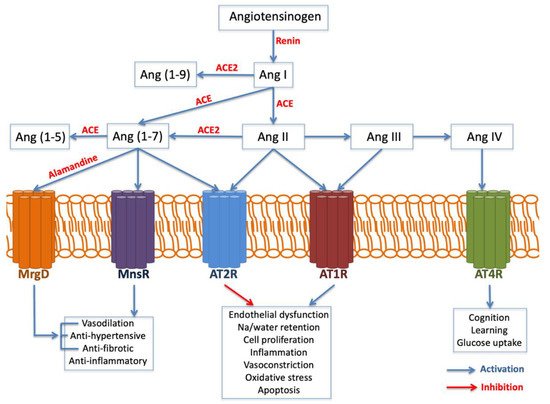
On the other hand, the angiotensin-converting enzyme (ACE) gene is placed on chromosome 17 band q23. Several studies have analyzed ACE insertion/deletion (I/D) polymorphism (rs4646994) Isbir and coworkers [72][97] found a relationship between the ACE D allele and an augmented susceptibility of postoperative AKI after coronary artery bypass graft surgery. Conversely, du Cheyron and colleagues [73][100] identified that I/I genotype is related to a higher vulnerability to AKI and RRT.
eNOS is the principal responsible for the vascular generation of nitric oxide, hence its variants might play a crucial role in the pathogenesis of endothelial dysfunction. Popov et al. demonstrated that T-786C eNOS polymorphism may predispose renal dysfunction and increase the incidence of dialysis following cardiac surgery [74][102]. Likewise, Stafford and coworkers showed that 894T eNOS polymorphism was associated with cardiac surgery-associated AKI (CSA-AKI) in Caucasians, as this variant may increment vascular tone and contribute to medullary ischemia [71][67].
Cytochrome b245 is involved in phagocytosis. While the A allele of the rs8854 polymorphism in the Cytochrome b245 α subunit (CYBA) gene was associated with less renal replacement therapy and hospital death compared to the GG genotype, the haplotype A-A-G-G of polymorphisms rs4782390, rs4673, rs3794624 and rs8854 was related to a higher vulnerability to this outcome [75][103].
Kallikreins belong to a family of serine proteases with diverse physiological functions. Kallikrein-1 (KLK1), one of 15 kallikrein family members, is the principal kallikrein expressed in the kidney and is implicated in both renal function and blood pressure control via vasodilatory and natriuretic results [76][104].
KLK1 is encoded by the KLK1 gene, which is located on chromosome 19q13.3 [76][104]. Polymorphisms at the promoter region have been identified and associated with CKD [77][78][105,106]. Susantitaphong and colleagues found that the I and G alleles of the KLK1 promoter polymorphism were related to an increased risk for AKI severity, including a doubled increase in serum creatinine, oliguria and RRT [76][104].
Perianayagam et al. [79][107] found that the rs4673 variant in the gene that encodes the NADPH oxidase p22phox subunit at position +242 is related to RRT and death among patients with AKI.
PH domain and leucine-rich repeat protein phosphatase 2 (PHLPP2) is a phosphatase essential for the control of PKC isoforms and Akt kinases [80][81][108,109]. Akt kinases, known as prosurvival kinases, regulate the equilibrium between cell survival and apoptosis, aside from proliferation and cellular quiescence. The rs78064607 polymorphism located in the PHLPP2 gene was the only SNP identified by Westphal et al. [82][110] in their genome-wide study with increased risk for AKI.
AKI produces the synthesis and release of catecholaminergic hormones as a response to any acute physiologic stress. As such, this pathway may play a relevant role in the etiology, pathogenesis, evolution and outcome of the disease.
The catechol-O-methyltransferase gene (COMT) encodes the COMT enzyme, which degrades catecholamines and contributes to vasodilatory shock and AKI [83][111]. Albert et al. and Haase-Flelitz et al. found that the low activity (L) rs4680 polymorphism or LL genotype is related to CSA-AKI, more furosemide administration and RRT [83][84][111,112]; however, this association was discarded by Kornet et al.
PNMT is the final product of the catecholaminergic pathway and converts noradrenaline to adrenaline. While the gene rs5638 + 1543 G allele is related to an augmented predisposition for AKI, and the genotype +1543 G/A is related to oliguria, the PNMT rs876493-161 A allele is associated with diminished mortality and less circulatory collapse [85][114].
GRM7 encodes a protein G-coupled receptor and is one of the group III metabotropic glutamate receptors, which are linked to the inhibition of the AMPc cascade. LMCD1-AS1 is a recognized oncogene and exerts a proliferation function.
The intergenic regionGRM7|LMCD-AS1located at chromosome 3p21.6 was detected to be highly related to CSA-AKI by Stafford-Smith et al., although no direct functional role was currently attributed to this intergenic region [86][115]. Future studies are needed to confirm this finding.
The SIK family members, including SIK3, are serine/threonine kinases that belong to the AMP-activated protein kinase (AMPK) family [87][88][116,117]. As their activity is upregulated in various cancers, they might play a key role in tumor appearance and progression. Polymorphism rs625145 in the SIK3 gene was correlated to elevated risk for AKI in patients with septic shock [89][118].
Hedgehog signaling is one of the crucial regulators of cell differentiation and has been deeply studied in the context of cancer. It also plays a key role in immune activation and inflammation, and its expression is upregulated while damaged organs are being healed. A key negative regulator of this signaling process is the suppressor of fused homolog (SUFU).
The polymorphisms rs10786691, rs12414407, rs10748825 and rs7078511 in the SUFU gene have been associated with renal performance in ICU patients with Enterobacteriaceae sepsis [90][119].
Transducer and activator of transcription 3 (STAT3) is a controller of T-helper 17 (Th17 cells) differentiation and function. Furthermore, STAT3 regulates and responds to various cytokines, including IL-1β, IL-10, IL-6, IL-8, IL-11, IL-17, IL-21 and IL-23 [91][120].
The STAT3 rs1053004 polymorphism was significantly related to a lower vulnerability of CSA-AKI in an older Iranian population [92][121].
While no association with AKI was found, Popov identified that the EPO gene rs1617640 T/G-polymorphism TT genotype was related to a higher creatine phosphokinase-MB (CPK-MB) and RRT requirement [93][122].
The SP-D gene is located in chromosome 10q22.2-q23.1 and is expressed in kidney tubules [94][95][123,124]. [96][125] demonstrated that patients with SP-D-11Thr/Thr genotype were more prone to AKI compared to those with other SP-D genotypes in a Chinese population. They showed that the more severely damaged the renal epithelial cells, the more SP-D protein will escape into the bloodstream, explaining the relationship between SP-D levels and AKI severity. In fact, greater serum SP-D levels were related to adverse clinical outcomes, such as higher AKI stage, longer RRT and increased mortality [96][125].
Adiponectin is a multifaceted cytokine that has a major role in the adjustment of energy metabolism and inflammatory response [97][98][126,127]. While initial reports informed that they are exclusively encountered in adipocytes [99][128], recent research has demonstrated that they are also produced by lymphocytes [100][129], macrophages and endothelial and epithelial cells [101][130]. Bloodstream adiponectin levels are higher in patients with chronic kidney disease (CKD), and elevated levels of adiponectin predict increased cardiovascular and all-cause mortality and CKD progression [102][131].
Jin et al. [103][132] showed that adiponectin plays a pivotal role in the pathogenesis of acute renal ischemia/reperfusion injury and may be a potential therapeutic target.
Apolipoprotein E is a protein implicated in lipid homeostasis, tissue restoration and immune response. The gene is located on chromosome 19q13.2 [104][44]. The polymorphism rs7412 of apolipoprotein E (APOE) and the non-e4 allele of polymorphism rs429358 are related to a greater peak serum creatinine [105][106][133,134] and an increased risk of postoperative AKI [72][106][97,134].
Variations in apolipoprotein L1, encoded by the APOL1 gene, are predisposing factors for focal segmental glomerulosclerosis, chronic renal and end-stage renal disease in African Americans [107][108][135,136], which may be related to renal blood flow impairment.
IRF2 controls the expression of the kidney disease risk gene APOL1. Zhao and colleagues found that rs62341639 and rs62341657 polymorphism on chromosome 4 near the APOL1 regulator IRF2 and rs9617814 and rs10854554 polymorphism on chromosome 22 close to the acute kidney injury-related gene TBX1 were associated with AKI development but without genome-wide significance [109][137]. TBX1 is a T-box transcription factor implicated in embryonic renal development, which was associated with the stimulation of TGF-β and with renal damage in a gentamicin-induced AKI study [110][138].
Hypoxia-inducible factor-1-α controls the cell response to hypoxic conditions. In 2009, Kolyada et al. [111][139] found that the T allele of the rs11549465 variant located on the transcription factor of the hypoxia-inducible factor-1alpha (HIF-1α) gene is associated with the increased need for RRT.
Myeloperoxidase is a lysosomal enzyme involved in oxidative stress and helps granulocytes destroy pathogens. In 2012, Perianayagam found that rs2243828, rs2071409, (rs2759) and rs7208693 polymorphisms of the MPO gene were associated with lower urine output, more dialysis requirement and higher in-hospital mortality [112][140].
Oxidative stress plays a major role in the pathogenesis of multiple conditions, and AKI is not an exception. The balance between the formation of reactive oxygen species and antioxidants is determinant. The T allele of the rs769217 variant of the antioxidant defense enzyme catalase (CAT) gene was associated with hospital morbidity and death among AKI patients in a Turkish population [113][141].
BCL2 has a major part in apoptosis signaling as an antiapoptosis protein [114][142]. Carriers having the minor alleles of rs8094315 and rs12457893 polymorphism on BCL2 had a declined predisposition for developing AKI [89][118].
SERPIN clade (SERPINA4) gene encodes kallistatin, which has vasodilatory, antioxidant, anti-inflammatory, and antiapoptotic properties [115][143]. Kallistatin suppresses TNF-α induced-apoptosis in in vitro endothelial cells [116][144]. Thus, polymorphism rs2093266 in the SERPINA4 gene was found to protect patients with septic shock against AKI [89][118].
Bardet–Biedl syndrome 9 (BBS9) gene plays a fundamental role in the control of cilia length through the adjustment of actin cytoskeleton polymerization [117][145]. Stafford-Smith and colleagues [86][115] in their GWAS identified a relationship of polymorphism rs10262995 in BBS9 with CSA-AKI. did not find this association [118][47].
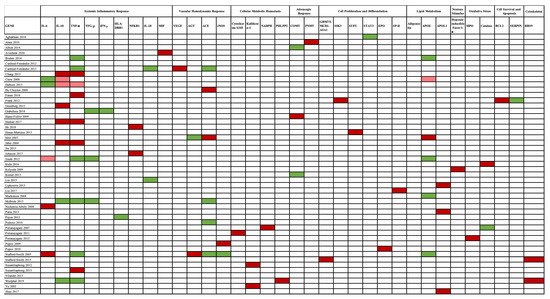
The association between gene studies and AKI susceptibility is depicted in Figure 4.
3. Conclusions
The overview of AKI pathogenesis suggests that various genes act collectively, generating either a favorable or harmful environment of pro- and anti-inflammatory cytokines, which determines the intensity of tissue damage. From the review, the most relevant genes are those related to the systemic inflammatory response, especially TNF-α, which illustrates the huge impact that this process has on the etiology and pathophysiology intricacy of this disease. Genetic variants may influence the kidney response to an injury, deciding whether a patient moves towards a more serious condition or recovery. Thus, genetic variety provides a useful rational approach as clinical risk factors, unfolding partially of the global risk.
As it can help identify susceptible patients based on genotype patterns with the aim to prevent or ameliorate kidney damage. A further understanding of these genetic variants can serve to develop a substantial improvement to tackle this disease through the development of novel risk stratification scores and new genetic specific targets. For instance, even though novel biomarkers, such as neutrophil gelatinase-associated lipocalin (NGAL), KIM-1, tissue inhibitors of metalloproteinases 1 (TIMP-1) and insulin-like growth factor binding protein 2 Thus, the knowledge of genetic determinants could benefit patient management as it can lend a hand to the creation of a genetic risk stratification tool with the development of genetic susceptibility biomarkers.