Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Camila Xu and Version 3 by Camila Xu.
Isoquinolones (isoquinolin-1(2H)-ones): the atom- and step-economic syntheses.
- alkyne
- annulation
- isoquinolone
1. Introduction
1. Introduction
Annulation protocols with unsaturated hydrocarbons are important and efficient methods for syntheses of cyclic compounds with high atom-utilization and step-economy, which have been extensively investigated in synthetic chemistry [1,2,3,4,5,6,7,8,9,10]. Isoquinolones are not only interesting and important nitrogen-heterocyclic compounds showing versatile biological and physiological activities but also useful synthetic blocks [11,12,13,14]. Several efficient approaches are available for the syntheses of isoquinolones, and the reported early works include the reactions of pyrroline-2,3-diones with benzyne [15], using dilithiated N-propenyl-ortho-toluamides [16], dilithiated 2-methyl-N-aryl-benzamides [17] as intermediates, and multi-step including SNAr reaction [18].
Over the past decades, isoquinolone ring construction through inter-/intramolecular annulation protocols of unsaturated hydrocarbons with nitrogen-containing reaction partners have been rapidly developed by the activation of aryl C–H and aryl C–X (X = halogen) bonds, as well as N–H or N–O bonds. This short review provides an overview of the recent advances on this theme.
Annulation protocols with unsaturated hydrocarbons are important and efficient methods for syntheses of cyclic compounds with high atom-utilization and step-economy, which have been extensively investigated in synthetic chemistry [1,2,3,4,5,6,7,8,9,10]. Isoquinolones are not only interesting and important nitrogen-heterocyclic compounds showing versatile biological and physiological activities but also useful synthetic blocks [11,12,13,14]. Several efficient approaches are available for the syntheses of isoquinolones, and the reported early works include the reactions of pyrroline-2,3-diones with benzyne [15], using dilithiated N-propenyl-ortho-toluamides [16], dilithiated 2-methyl-N-aryl-benzamides [17] as intermediates, and multi-step including SNAr reaction [18].
Over the past decades, isoquinolone ring construction through inter-/intramolecular annulation protocols of unsaturated hydrocarbons with nitrogen-containing reaction partners have been rapidly developed by the activation of aryl C–H and aryl C–X (X = halogen) bonds, as well as N–H or N–O bonds.
2. Isoquinolone Formation via [4 + 2] Intermolecular Annulations and Analogous Reactions
The cyclocondensation of benzamide derivatives and functionalized arenes with alkynes or C2 synthons have been recently made great progress for the efficient access to isoquinolones via a [4 + 2] annulation manner. The computational studies on the transition-metal-catalyzed aryl C–H oxidative cyclocondensation of benzamides with alkynes affording isoquinolones have also been reported [19].
2.1. [4 + 2]. Intermolecular Annulations via Aryl C–X/N–H Activation
The [4 + 2] intermolecular annulations of ortho-halobenzamides with alkynes via activation of aryl C–X (X = halogen)/N–H bond affording N-substituted isoquinolones have been extensively studied in the presence of nickel complexes under different conditions (Scheme 1). The general proposed mechanism includes the oxidative addition of orhto-halobenzamide to nickel (0) generated in situ in the presence of base leading to the formation of nickelacycle, coordinative insertion of alkyne into the nickelacycle giving possible two seven-membered ring nickelacycle intermediates, followed by reductive elimination affording isoquinolinone [20].
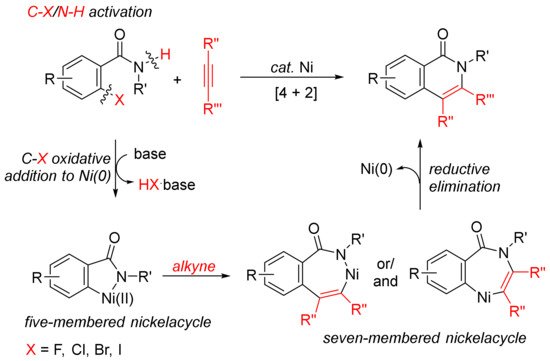
Scheme 1. Isoquinolone formation by [4 + 2] intermolecular annulation of ortho-halobenzamides with alkynes via C–X/N–H activation.
With the use of air-stable [Ni(dppe)Br2] as catalyst, in the presence of Zn and Et3N, the reactions of ortho-halobenzamide with internal alkynes afford N-substituted isoquinolone derivatives (R’ = alkyl, allyl, benzyl, aryl) in good to high yields [20]. In the cases of unsymmetric internal alkynes and terminal alkynes used, the cyclocondensation shows regioselective manner, and the catalytic procedure can be used for the synthesis of isoquinolinone alkaloid natural products.
Ni(dppp)Cl2 can also catalyze the annulation of ortho-iodobenzamides with alkynes in the presence of Et3N in MeCN by selective cleavage of C–I/N–H bond [21]. Very recently, Ni(cod)2/KOtBu-catalyzed formation of highly substituted isoquinolones through cyclocondensation of ortho-fluorobenzamides with internal alkynes via C–F/N–H activation has also been reported [22].
With the use of ortho-haloarylamidines as substrates, in the presence of H2O and zinc, Ni(dppp)Cl2-catalyzed annulations with terminal and internal alkynes affords N-aryl isoquinolones in good to high yields (Scheme 2) [23].
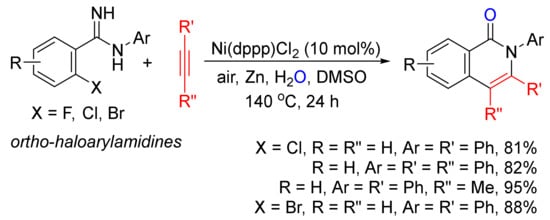
Scheme 2. Nickel-catalyzed annulation of ortho-haloarylamidines with alkynes affording N-aryl isoquinolones.
β-Keto esters are used as a C2 synthon in the formation of isoquinolones via [4 + 2] cyclocondensation with ortho-halobenzamides [24]. As shown in Scheme 3, CuI-catalyzed reactions of ortho-chloro, -bromo-, and -iodobenzamides or ortho-chloronicotinamide with β-keto esters in the presence of Cs2CO3 without addition of any ligand or additive afford 3,4-disubstituted N–H isoquinolone derivatives in moderate to high yields.
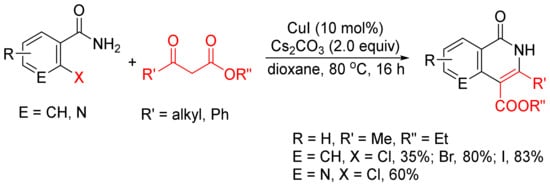
Scheme 3. CuI-catalyzed synthesis of 3,4-disubstituted isoquinolones.
In addition, as shown in Scheme 4, in the presence of Pd(OAc)2 and PtBu3HBF4, norbornadiene can also be used as an acetylene equivalent in the synthesis of 3,4-unsubstituted N-benzyl isoquinolones via the annulation with ortho-chlorobenzamides [25]. A Cu(OAc)2.H2O-catalyzed cyclocoupling reaction of N-substituted ortho-iodo-benzamides with malononitrile afforded N-substituted-3-amino-4-cyano-isoquinolones in good to high yields, and malononitrile is used as C2 synthon [26].
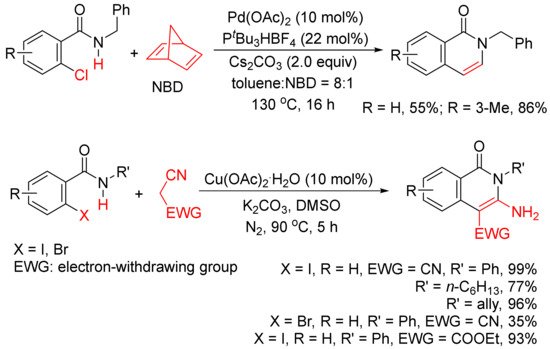
Scheme 4. Isoquinolone formation by cyclocoupling reactions of ortho-halobenzamides with norbornadiene or malononitrile.
The formation of functionalized isoquinolones by copper-catalyzed cyclocondensation of ketones with ortho-halobenzamides [27] or ortho-halobenzonitriles [28] through SNAr reaction giving α-aryl ketones as the intermediates have also been reported (Scheme 5).
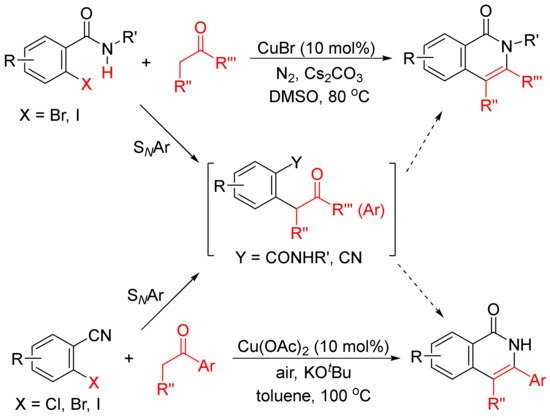
Scheme 5. Copper-catalyzed isoquinolone formation including SNAr reaction.
2.2. [4 + 2]. Intermolecular Annulations via Aryl C–H/N–O Activation Access to N–H Isoquinolones
Transition-metal-catalyzed annulations of alkynes with N-alkoxybenzamides by C–H/N–O bond cleavages with dealkoxylation reaction have been extensively investigated for the formation of N–H isoquinolones (Scheme 6).
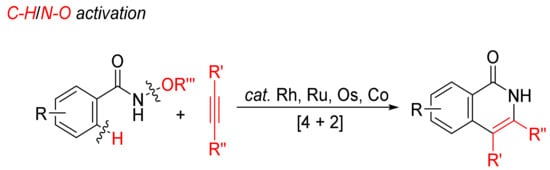
Scheme 6. Isoquinolone formation by [4 + 2] intermolecular annulation of N-alkoxybenzamides with alkynes via C–H/N–O activation.
Guimond and coworkers first studied the cyclocondensation of N-alkoxybenzamides with internal alkynes catalyzed by [Cp*RhCl2]2/CsOAc in methanol to synthesize N–H isoquinolones via C–H/N–O bond activation in 2010, and the mechanism was also investigated in detail [29,30]. The group CONH(OMe) is used as an oxidizing directing group, and the transformation occurs under external oxidant-free conditions.
The same catalyst system was then applied in the intramolecular reaction of alkyne-tethered N-alkoxybenzamides leading to a general and facile synthesis of 3-hydroxyalkyl-substituted N–H isoquinolones as the major products, and one of them can be used in the synthesis of natural product of Rosettacin, which is a polycyclic-fused isoquinolone derivative (Scheme 7) [31].
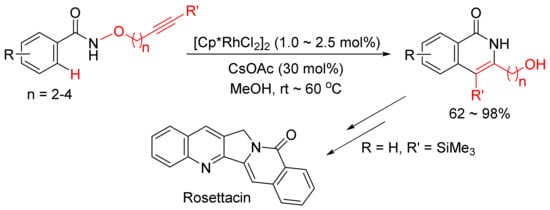
Scheme 7. Synthesis of Rosettacin.
Similar conditions were then applied by other groups in the [4 + 2] cyclocondensation of N-pivaloyloxy benzamides with vinyl acetate [32], cyclopropenes [33], and substituted allenes [34] to develop the synthesis of 3,4-unsubstituted N–H isoquinolones and 3- or/and 4-substituted N–H isoquinolones (Scheme 8). In the case of the vinyl acetate used, it emerges as the convenient acetylene equivalent.
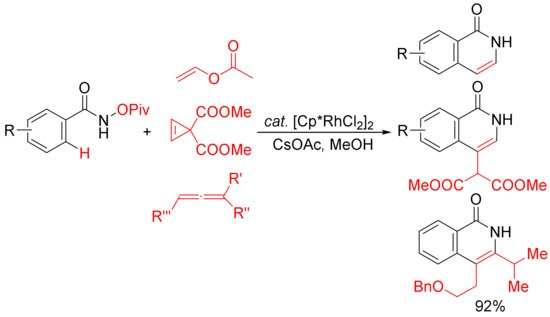
Scheme 8. Rh(III)-catalyzed C–H/N–O activation/annulation of N-OPiv benzamides with vinyl esters, cyclopropenes, and allenes affording N–H isoquinolones.
In addition, 3,4-unsubstituted N–H isoquinolones can also be synthesized by annulation of benzoic acid with N-vinyl formamide catalyzed by [Cp*RhCl2]2 in the presence of AgOAc and KHCO3 in benzonitrile at 80 °C [35].
Thereafter, catalysis of [Cp*RhCl2]2/Bu4NOAc in DCE [36], [RuCl2(p-cymene)]2/NaOAc in MeOH [37,38], [RuCl2(p-cymene)]2/KO2CMes in water [39], [RuCl2(p-cymene)]2/KO2CMes/Cu(OAc)2.H2O in CHCl3 [40], [Os(p-cymene)Cl2]2/NaOAc/HOAc in trifluoroethanol (TFE) [41], and Co(OAc)2.4H2O/AgOAc/PivOH in TFE [42] have been developed to synthesize N–H isoquinolones from the reactions of secondary N-alkoxybenzamides with internal alkynes via selective N–O bond cleavage. The use of free hydroxamic acids is also successful to give the corresponding isoquinolones [39].
In addition, [RuCl2(p-cymene)]2/NaOAc in MeOH can be applied in the reactions of N-methoxybenzamide with alkynyl bromides affording 3-methoxy-4-substituted N–H isoquinolinones with excellent chemoselectivity via C–H/N–O activation [43]. Recently, the same catalyst system has been applied in the intramolecular cyclo-isomerization of alkyne-tethered N-alkoxybenzamides to give 3,4-disubstituted N–H isoquinolones (Scheme 9) [44]. The experimental and theoretical studies indicate that the reaction to the inverse annulation follows the Ru(II)-Ru(IV)-Ru(II) pathway involving N–O bond cleavage prior to alkyne insertion.
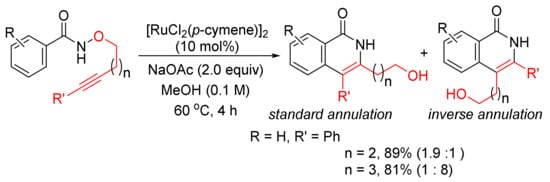
Scheme 9. Ruthenium-catalyzed cascade C–H activation/annulation of alkyne-tethered N-alkoxybenzamides.
Moreover, the functionalized arenes have also been used as the coupling partners in the [4 + 2] annulations with alkynes to construct isoquinolone ring via activation of aryl C–H and N–E (E = N, C, O, Cl, etc.) bonds. As shown in Scheme 10, with the use of [Cp*RhCl2]2 as catalyst, the annulations of alkynes with benzoyl hydrazines (Scheme 10 (a)) [45], with 1,4,2-dioxazol-5-ones occur at room temperature (Scheme 10 (b)) [46], with N-iminopyridinium ylides (Scheme 10 (c)) [47]. However, with the use of AgNTf2 as co-catalyst, the reactions of 2-aryloxazolines with alkynes afford N-substituted isoquinolones (Scheme 10 (d)) [48].
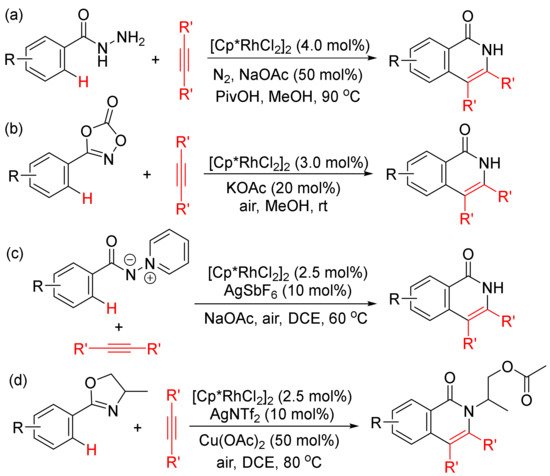
Scheme 10. Isoquinolone formation by rhodium (III)-catalyzed annulation of alkynes with different aryl substrates.
In the presence of KOAc, [Cp*Co(CO)I2]/AgOAc-catalyzed [4 + 2] annulation of N-chlorobenzamides with alkynes in TFE occurs at room temperature with selective cleavage of C–H/N–Cl bonds (Scheme 11) [49]. In the case of the use of terminal alkyne, the reactions occur with high regioselectivity to give 3-substituted N–H isoquinolones.
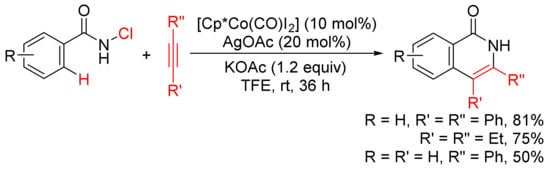
Scheme 11. N–H isoquinolone formation by cobalt-catalyzed cyclocondensation of N-chloroamides with alkynes.
2.3. [4 + 2]. Intermolecular Annulations via Aryl C–H/N–H Activation
More atom-economical procedures for the formation of isoquinolones is the oxidative [4 + 2] cyclocondensation of primary and secondary benzamides with internal alkynes with selective cleavages of C–H and N–H bonds (Scheme 12).
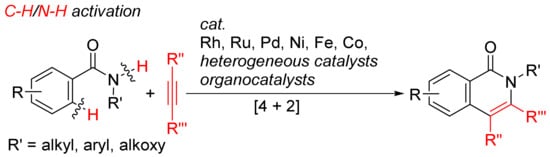
Scheme 12. Isoquinolone formation by [4 + 2] intermolecular annulation of benzamides with alkynes via C–H/N–H activation.
Miura and Satoh’s group reported the oxidative coupling of primary, secondary, and tertiary benzamides with internal alkynes catalyzed by rhodium complexes and with the use of Cu(OAc)2.2H2O as oxidant in 2010 [50]. As shown in Scheme 13, the reaction between primary benzamides with internal alkynes affords isoquinoline-fused polycyclic aromatics resulting from two incorporated molecules of alkynes. In the case of secondary benzamides, N-substituted isoquinolones are formed. Interestingly, the reaction of tertiary benzamides with internal alkynes under similar conditions produces 1-naphthalenecarboxamides through double aryl C–H bond activation (Scheme 14).
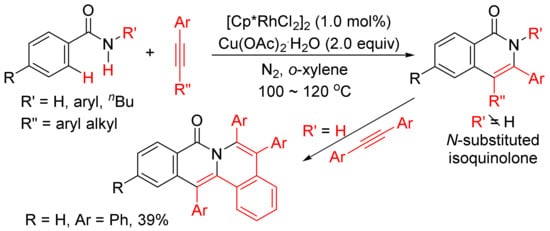
Scheme 13. Rhodium-catalyzed cyclocondensation of primary/secondary benzamides with internal alkynes affording isoquinolones and fused isoquinolone.
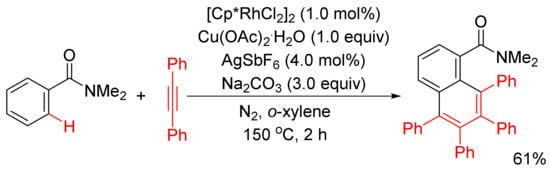
Scheme 14. Rhodium-catalyzed oxidative coupling of tertiary benzamides with internal alkynes.
In the same year, two similar works were also reported using [Cp*RhCl2]2 as catalyst to synthesize N-substituted isoquinolones by oxidative/cyclization of benzamides with internal alkynes via selective aryl C–H/N–H bonds cleavage [51,52]. Other rhodium catalysis of [Cp*Rh(MeCN)3]BF4/K2CO3 in water under air was then developed [53].
Although [RuCl2(p-cymene)]2-catalyzed annulations of N-alkoxybenzamides (R’ = alkoxy) with internal alkynes occur with dealkoxylation reactions via activation of N–O bond to give N–H isoquinolones [37,38,39,40], with the use of Cu(OAc)2·H2O as the terminal oxidant and t-AmOH as the solvent, the oxidative annulation of N-alkyl/arylbenzamides (R’ = alkyl, aryl) with internal alkynes gives efficient access to N-alkyl/aryl-substituted isoquinolones by highly selective cleavage of N–H bond [54,55].
In addition, when Pd(OAc)2 was used as a catalyst, in the presence of NaI.2H2O as additive, the methoxy substituent on nitrogen (R’ = alkoxy) was not cleaved during the catalysis under air, and the formation of N-methoxy isoquinolones had high chemoselectivity (Scheme 15) [56,57]. Under similar reaction conditions, using Cu(II) salts as the co-oxidants, a broad range of N-alkyl and N-aryl-substituted isoquinolones can also be obtained starting from N-alkyl/aryl benzamides, and N-C bonds are not cleaved either [58]. The mechanistic studies propose that the use of stoichiometric amount of Cu(II) salts as the key oxidant and air as the terminal oxidant are key for the regeneration of active Pd(II) species.
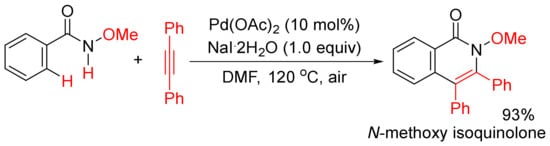
Scheme 15. N-Methoxy isoquinolone formation via highly chemoselective N–H activation.
A similar catalyst system was then applied in the oxidative annulation of N-methoxybenzamides with arynes and cyclooctynes via C–H/N–H activation for the synthesis of fused isoquinolones [59]. Very recently, the annulation of benzamides with arynes using palladium with photoredox dual catalysis has also been developed for the synthesis of benzo-fused isoquinolones [60].
The formation of N-alkyl-, aryl-, or alkoxy-substituted isoquinolones by the oxidative [4 + 2] cyclocondensation of secondary benzamides with internal alkynes with selective cleavages of C–H and N–H bonds has been achieved using other transition-metal catalyst systems, such as nickel [61,62], iron [63] complexes as catalysts, and heterogeneous catalyst of palladium-nanoparticles [64], Pd/C [65,66].
In the presence of NaOPiv·H2O, Co(OAc)2·4H2O/Ag2O-catalyzed decarboxylative C–H/N–H activation/annulation of N-PyO benzamides with alkynyl carboxylic acids under air in TFE also affords 3-substituted isoquinolones with high regioselectivity [67]. Similarly, 3-substituted isoquinolones can be obtained using Co(OAc)2·4H2O as catalyst from the reactions of N-(quinolin-8-yl)benzamides with alkynylsilanes with desilylation under oxygen in the same solvent, with the use of Mn(acac)2 and CsF [68].
In addition, the first metal-free annulation of N-methoxybenzamides with internal alkynes was reported by Antonchick’s group with the use of PhI as catalyst and AcOOH as oxidant in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at room temperature [69]. A mechanism involving the formation of hypervalent iodine reagent of (diacetoxyiodo)benzene (PIDA) is proposed. At the same time, this type of oxidative cycloaddition reaction can also occur using bis(trifluoracetoxy)iodobenzene as oxidant in the presence of trifluoroacetic acid [70].
Rh(III)-catalyzed annulation of benzoyl hydrazines and alkynes affords isoquinolones with deammoniation by cleavage of N-N bond (Scheme 10 (a)). However, [Ru(p-cymene)Cl2]2/AgSbF6-catalyzed reactions of N-(1H-pyrrolo [2,3-b]pyridin-1-yl)-benzamides with internal alkynes afford isoquinolone derivatives with remaining N-N bond untouched (Scheme 16) [71]. In this procedure, N-amino-7-azaindole is used as an efficient bidentate-directing group to promote the selective C–H bond activation.
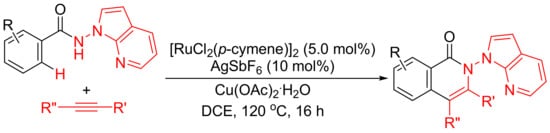
Scheme 16. Ruthenium-catalyzed isoquinolone formation by N-amino-7-azaindole-assisted C–H activation.
Instead of internal alkynes, the annulations of benzamides with other C2 synthons via C–H/N–H activation to construct isoquinolone ring have also been developed. For example, Glorius and coworkers developed a Rh(III)-catalyzed intermolecular [4 + 2] cyclocondensation of N-methoxybenzamides with α-halo and pseudohalo ketones to afford various 3-substituted N-methoxyisoquinolones in moderate to excellent yields (Scheme 17) [72]. The α-halo and pseudohalo ketones are utilized as oxidized alkyne equivalents to undergo the redox-neutral annulation, and through the formation of ortho-acylmethyl-benzamides as the intermediates. Recently, Ma and coworkers have also reported a Pd(OAc)2/L-alanine-catalyzed formation of 3-substituted N-(quinolin-8-yl)-isoquinolones with by N-(quinolin-8-yl)benzamides and α-bromo ketones in the presence of K2CO3 in t-AmOH [73].
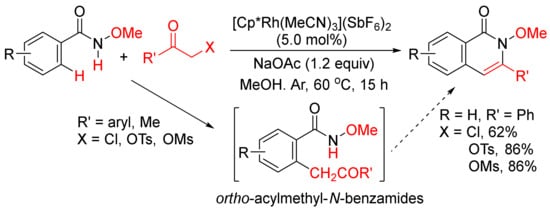
Scheme 17. 3-Substituted isoquinolone synthesis via Rh(III)-catalyzed intermolecular cyclocondensation of N-methoxybenzamides with α-halo and pseudohalo ketones.
With the use of same rhodium complex, 4-substituted isoquinolones can be obtained by a microwave-assisted annulation of N-methoxyamides with α-chloroaldehydes (Scheme 18) [74].
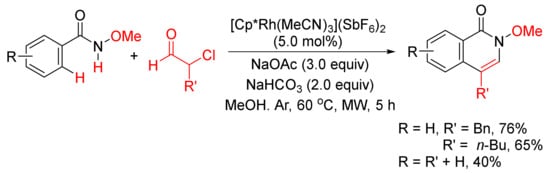
Scheme 18. 4-Substituted isoquinolone formation via microwave-assisted annulation of N-methoxyamides with α-chloroaldehydes.
Moreover, 3-Cyclohexylmethyl N-methoxyisoquinolone, which was published as only one example in literature [75], can be synthesized by the cyclocondensation of N-methoxyisoquinolone with the corresponding α-carbonyl sulfoxonium ylide catalyzed by rhodium in HFIP (Scheme 19). The detailed studies for the formation of 3-substituted isoquinolones through the reactions of N-methoxybenzamides and α-carbonyl sulfoxonium ylides catalyzed by Cp*Rh(MeCN)3(SbF6)2/Zn(OTf)2 in DCE were then reported by Li and coworkers [76].
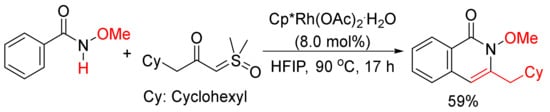
Scheme 19. Rhodium-catalyzed cyclocondensation of N-methoxyisoquinolone with α-carbonyl sulfoxonium ylide affording 3-substituted isoquinolone.
Very recently, epoxides as alkylating reagents and C2 synthon have been applied in Pd(OAc)2-catalyzed oxidative annulation of N-alkoxybenzamides to regioselectively synthesize 3-substituted isoquinolones in HFIP solvent (Scheme 20) [77]. The present methodology has been successfully employed in the total syntheses of rupreschstyril, siamine, and cassiarin A (
2. Isoquinolone Formation via [4 + 2] Intermolecular Annulations and Analogous Reactions
The cyclocondensation of benzamide derivatives and functionalized arenes with alkynes or C2 synthons have been recently made great progress for the efficient access to isoquinolones via a [4 + 2] annulation manner. The computational studies on the transition-metal-catalyzed aryl C–H oxidative cyclocondensation of benzamides with alkynes affording isoquinolones have also been reported [19].
2.1. [4 + 2]. Intermolecular Annulations via Aryl C–X/N–H Activation
The [4 + 2] intermolecular annulations of ortho-halobenzamides with alkynes via activation of aryl C–X (X = halogen)/N–H bond affording N-substituted isoquinolones have been extensively studied in the presence of nickel complexes under different conditions (Scheme 1). The general proposed mechanism includes the oxidative addition of orhto-halobenzamide to nickel (0) generated in situ in the presence of base leading to the formation of nickelacycle, coordinative insertion of alkyne into the nickelacycle giving possible two seven-membered ring nickelacycle intermediates, followed by reductive elimination affording isoquinolinone [20].
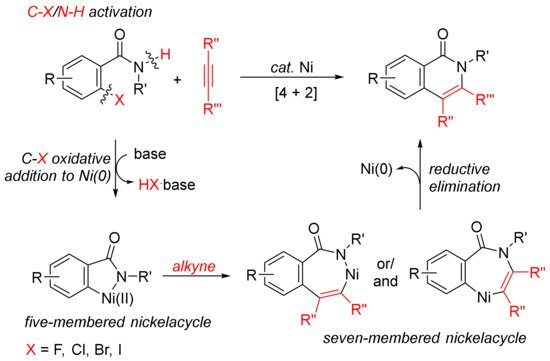
Scheme 1. Isoquinolone formation by [4 + 2] intermolecular annulation of ortho-halobenzamides with alkynes via C–X/N–H activation.
With the use of air-stable [Ni(dppe)Br2] as catalyst, in the presence of Zn and Et3N, the reactions of ortho-halobenzamide with internal alkynes afford N-substituted isoquinolone derivatives (R’ = alkyl, allyl, benzyl, aryl) in good to high yields [20]. In the cases of unsymmetric internal alkynes and terminal alkynes used, the cyclocondensation shows regioselective manner, and the catalytic procedure can be used for the synthesis of isoquinolinone alkaloid natural products.
Ni(dppp)Cl2 can also catalyze the annulation of ortho-iodobenzamides with alkynes in the presence of Et3N in MeCN by selective cleavage of C–I/N–H bond [21]. Very recently, Ni(cod)2/KOtBu-catalyzed formation of highly substituted isoquinolones through cyclocondensation of ortho-fluorobenzamides with internal alkynes via C–F/N–H activation has also been reported [22].
With the use of ortho-haloarylamidines as substrates, in the presence of H2O and zinc, Ni(dppp)Cl2-catalyzed annulations with terminal and internal alkynes affords N-aryl isoquinolones in good to high yields (Scheme 2) [23].
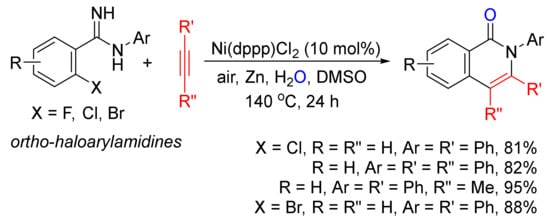
Scheme 2. Nickel-catalyzed annulation of ortho-haloarylamidines with alkynes affording N-aryl isoquinolones.
β-Keto esters are used as a C2 synthon in the formation of isoquinolones via [4 + 2] cyclocondensation with ortho-halobenzamides [24]. As shown in Scheme 3, CuI-catalyzed reactions of ortho-chloro, -bromo-, and -iodobenzamides or ortho-chloronicotinamide with β-keto esters in the presence of Cs2CO3 without addition of any ligand or additive afford 3,4-disubstituted N–H isoquinolone derivatives in moderate to high yields.
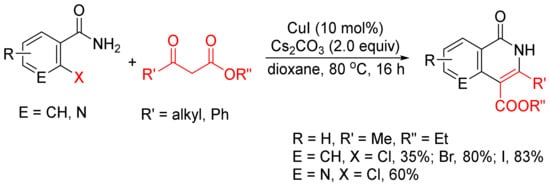
Scheme 3. CuI-catalyzed synthesis of 3,4-disubstituted isoquinolones.
In addition, as shown in Scheme 4, in the presence of Pd(OAc)2 and PtBu3HBF4, norbornadiene can also be used as an acetylene equivalent in the synthesis of 3,4-unsubstituted N-benzyl isoquinolones via the annulation with ortho-chlorobenzamides [25]. A Cu(OAc)2.H2O-catalyzed cyclocoupling reaction of N-substituted ortho-iodo-benzamides with malononitrile afforded N-substituted-3-amino-4-cyano-isoquinolones in good to high yields, and malononitrile is used as C2 synthon [26].
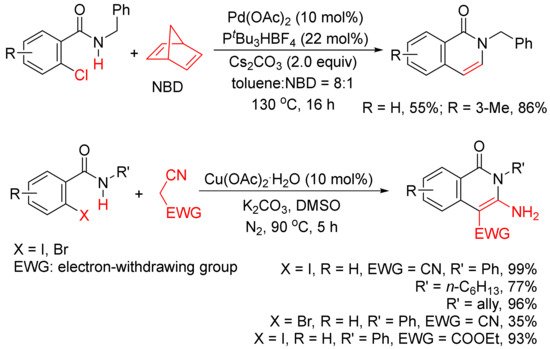
Scheme 4. Isoquinolone formation by cyclocoupling reactions of ortho-halobenzamides with norbornadiene or malononitrile.
The formation of functionalized isoquinolones by copper-catalyzed cyclocondensation of ketones with ortho-halobenzamides [27] or ortho-halobenzonitriles [28] through SNAr reaction giving α-aryl ketones as the intermediates have also been reported (Scheme 5).
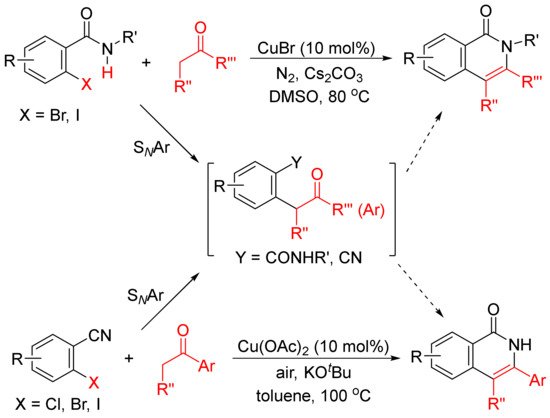
Scheme 5. Copper-catalyzed isoquinolone formation including SNAr reaction.
2.2. [4 + 2]. Intermolecular Annulations via Aryl C–H/N–O Activation Access to N–H Isoquinolones
Transition-metal-catalyzed annulations of alkynes with N-alkoxybenzamides by C–H/N–O bond cleavages with dealkoxylation reaction have been extensively investigated for the formation of N–H isoquinolones (Scheme 6).
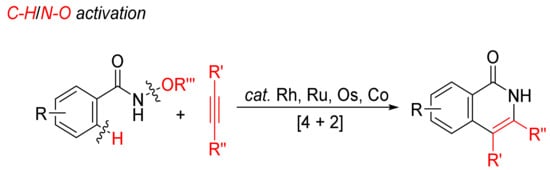
Scheme 6. Isoquinolone formation by [4 + 2] intermolecular annulation of N-alkoxybenzamides with alkynes via C–H/N–O activation.
Guimond and coworkers first studied the cyclocondensation of N-alkoxybenzamides with internal alkynes catalyzed by [Cp*RhCl2]2/CsOAc in methanol to synthesize N–H isoquinolones via C–H/N–O bond activation in 2010, and the mechanism was also investigated in detail [29,30]. The group CONH(OMe) is used as an oxidizing directing group, and the transformation occurs under external oxidant-free conditions.
The same catalyst system was then applied in the intramolecular reaction of alkyne-tethered N-alkoxybenzamides leading to a general and facile synthesis of 3-hydroxyalkyl-substituted N–H isoquinolones as the major products, and one of them can be used in the synthesis of natural product of Rosettacin, which is a polycyclic-fused isoquinolone derivative (Scheme 7) [31].
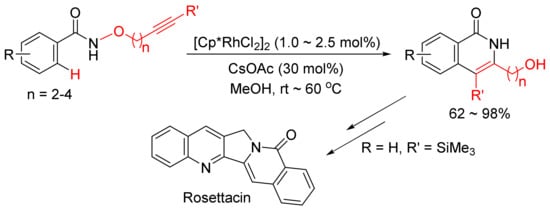
Scheme 7. Synthesis of Rosettacin.
Similar conditions were then applied by other groups in the [4 + 2] cyclocondensation of N-pivaloyloxy benzamides with vinyl acetate [32], cyclopropenes [33], and substituted allenes [34] to develop the synthesis of 3,4-unsubstituted N–H isoquinolones and 3- or/and 4-substituted N–H isoquinolones (Scheme 8). In the case of the vinyl acetate used, it emerges as the convenient acetylene equivalent.
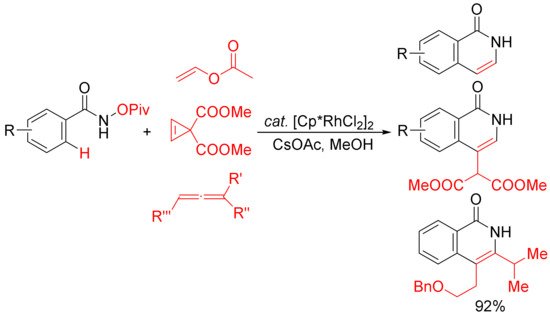
Scheme 8. Rh(III)-catalyzed C–H/N–O activation/annulation of N-OPiv benzamides with vinyl esters, cyclopropenes, and allenes affording N–H isoquinolones.
In addition, 3,4-unsubstituted N–H isoquinolones can also be synthesized by annulation of benzoic acid with N-vinyl formamide catalyzed by [Cp*RhCl2]2 in the presence of AgOAc and KHCO3 in benzonitrile at 80 °C [35].
Thereafter, catalysis of [Cp*RhCl2]2/Bu4NOAc in DCE [36], [RuCl2(p-cymene)]2/NaOAc in MeOH [37,38], [RuCl2(p-cymene)]2/KO2CMes in water [39], [RuCl2(p-cymene)]2/KO2CMes/Cu(OAc)2.H2O in CHCl3 [40], [Os(p-cymene)Cl2]2/NaOAc/HOAc in trifluoroethanol (TFE) [41], and Co(OAc)2.4H2O/AgOAc/PivOH in TFE [42] have been developed to synthesize N–H isoquinolones from the reactions of secondary N-alkoxybenzamides with internal alkynes via selective N–O bond cleavage. The use of free hydroxamic acids is also successful to give the corresponding isoquinolones [39].
In addition, [RuCl2(p-cymene)]2/NaOAc in MeOH can be applied in the reactions of N-methoxybenzamide with alkynyl bromides affording 3-methoxy-4-substituted N–H isoquinolinones with excellent chemoselectivity via C–H/N–O activation [43]. Recently, the same catalyst system has been applied in the intramolecular cyclo-isomerization of alkyne-tethered N-alkoxybenzamides to give 3,4-disubstituted N–H isoquinolones (Scheme 9) [44]. The experimental and theoretical studies indicate that the reaction to the inverse annulation follows the Ru(II)-Ru(IV)-Ru(II) pathway involving N–O bond cleavage prior to alkyne insertion.
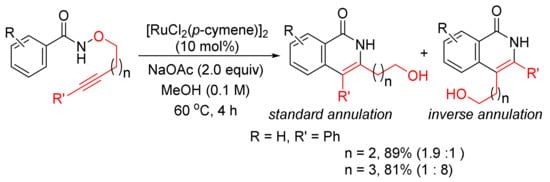
Scheme 9. Ruthenium-catalyzed cascade C–H activation/annulation of alkyne-tethered N-alkoxybenzamides.
Moreover, the functionalized arenes have also been used as the coupling partners in the [4 + 2] annulations with alkynes to construct isoquinolone ring via activation of aryl C–H and N–E (E = N, C, O, Cl, etc.) bonds. As shown in Scheme 10, with the use of [Cp*RhCl2]2 as catalyst, the annulations of alkynes with benzoyl hydrazines (Scheme 10 (a)) [45], with 1,4,2-dioxazol-5-ones occur at room temperature (Scheme 10 (b)) [46], with N-iminopyridinium ylides (Scheme 10 (c)) [47]. However, with the use of AgNTf2 as co-catalyst, the reactions of 2-aryloxazolines with alkynes afford N-substituted isoquinolones (Scheme 10 (d)) [48].
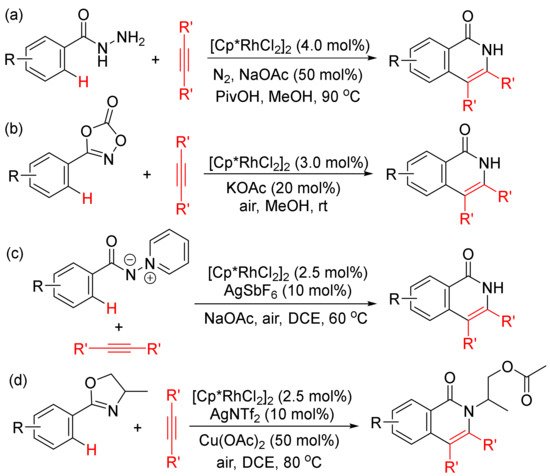
Scheme 10. Isoquinolone formation by rhodium (III)-catalyzed annulation of alkynes with different aryl substrates.
In the presence of KOAc, [Cp*Co(CO)I2]/AgOAc-catalyzed [4 + 2] annulation of N-chlorobenzamides with alkynes in TFE occurs at room temperature with selective cleavage of C–H/N–Cl bonds (Scheme 11) [49]. In the case of the use of terminal alkyne, the reactions occur with high regioselectivity to give 3-substituted N–H isoquinolones.
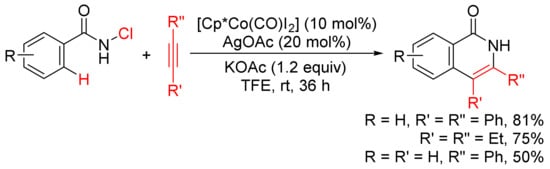
Scheme 11. N–H isoquinolone formation by cobalt-catalyzed cyclocondensation of N-chloroamides with alkynes.
2.3. [4 + 2]. Intermolecular Annulations via Aryl C–H/N–H Activation
More atom-economical procedures for the formation of isoquinolones is the oxidative [4 + 2] cyclocondensation of primary and secondary benzamides with internal alkynes with selective cleavages of C–H and N–H bonds (Scheme 12).
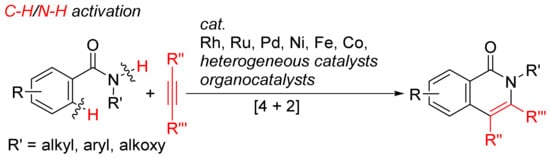
Scheme 12. Isoquinolone formation by [4 + 2] intermolecular annulation of benzamides with alkynes via C–H/N–H activation.
Miura and Satoh’s group reported the oxidative coupling of primary, secondary, and tertiary benzamides with internal alkynes catalyzed by rhodium complexes and with the use of Cu(OAc)2.2H2O as oxidant in 2010 [50]. As shown in Scheme 13, the reaction between primary benzamides with internal alkynes affords isoquinoline-fused polycyclic aromatics resulting from two incorporated molecules of alkynes. In the case of secondary benzamides, N-substituted isoquinolones are formed. Interestingly, the reaction of tertiary benzamides with internal alkynes under similar conditions produces 1-naphthalenecarboxamides through double aryl C–H bond activation (Scheme 14).
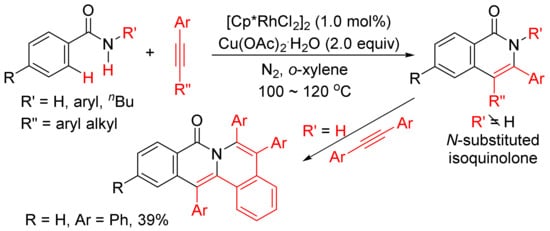
Scheme 13. Rhodium-catalyzed cyclocondensation of primary/secondary benzamides with internal alkynes affording isoquinolones and fused isoquinolone.
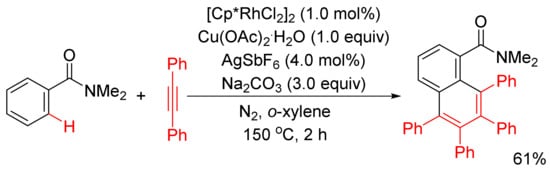
Scheme 14. Rhodium-catalyzed oxidative coupling of tertiary benzamides with internal alkynes.
In the same year, two similar works were also reported using [Cp*RhCl2]2 as catalyst to synthesize N-substituted isoquinolones by oxidative/cyclization of benzamides with internal alkynes via selective aryl C–H/N–H bonds cleavage [51,52]. Other rhodium catalysis of [Cp*Rh(MeCN)3]BF4/K2CO3 in water under air was then developed [53].
Although [RuCl2(p-cymene)]2-catalyzed annulations of N-alkoxybenzamides (R’ = alkoxy) with internal alkynes occur with dealkoxylation reactions via activation of N–O bond to give N–H isoquinolones [37,38,39,40], with the use of Cu(OAc)2·H2O as the terminal oxidant and t-AmOH as the solvent, the oxidative annulation of N-alkyl/arylbenzamides (R’ = alkyl, aryl) with internal alkynes gives efficient access to N-alkyl/aryl-substituted isoquinolones by highly selective cleavage of N–H bond [54,55].
In addition, when Pd(OAc)2 was used as a catalyst, in the presence of NaI.2H2O as additive, the methoxy substituent on nitrogen (R’ = alkoxy) was not cleaved during the catalysis under air, and the formation of N-methoxy isoquinolones had high chemoselectivity (Scheme 15) [56,57]. Under similar reaction conditions, using Cu(II) salts as the co-oxidants, a broad range of N-alkyl and N-aryl-substituted isoquinolones can also be obtained starting from N-alkyl/aryl benzamides, and N-C bonds are not cleaved either [58]. The mechanistic studies propose that the use of stoichiometric amount of Cu(II) salts as the key oxidant and air as the terminal oxidant are key for the regeneration of active Pd(II) species.
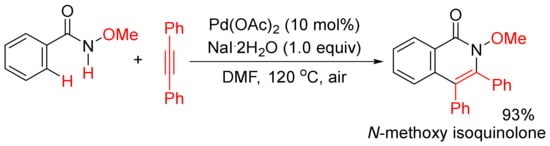
Scheme 15. N-Methoxy isoquinolone formation via highly chemoselective N–H activation.
A similar catalyst system was then applied in the oxidative annulation of N-methoxybenzamides with arynes and cyclooctynes via C–H/N–H activation for the synthesis of fused isoquinolones [59]. Very recently, the annulation of benzamides with arynes using palladium with photoredox dual catalysis has also been developed for the synthesis of benzo-fused isoquinolones [60].
The formation of N-alkyl-, aryl-, or alkoxy-substituted isoquinolones by the oxidative [4 + 2] cyclocondensation of secondary benzamides with internal alkynes with selective cleavages of C–H and N–H bonds has been achieved using other transition-metal catalyst systems, such as nickel [61,62], iron [63] complexes as catalysts, and heterogeneous catalyst of palladium-nanoparticles [64], Pd/C [65,66].
In the presence of NaOPiv·H2O, Co(OAc)2·4H2O/Ag2O-catalyzed decarboxylative C–H/N–H activation/annulation of N-PyO benzamides with alkynyl carboxylic acids under air in TFE also affords 3-substituted isoquinolones with high regioselectivity [67]. Similarly, 3-substituted isoquinolones can be obtained using Co(OAc)2·4H2O as catalyst from the reactions of N-(quinolin-8-yl)benzamides with alkynylsilanes with desilylation under oxygen in the same solvent, with the use of Mn(acac)2 and CsF [68].
In addition, the first metal-free annulation of N-methoxybenzamides with internal alkynes was reported by Antonchick’s group with the use of PhI as catalyst and AcOOH as oxidant in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at room temperature [69]. A mechanism involving the formation of hypervalent iodine reagent of (diacetoxyiodo)benzene (PIDA) is proposed. At the same time, this type of oxidative cycloaddition reaction can also occur using bis(trifluoracetoxy)iodobenzene as oxidant in the presence of trifluoroacetic acid [70].
Rh(III)-catalyzed annulation of benzoyl hydrazines and alkynes affords isoquinolones with deammoniation by cleavage of N-N bond (Scheme 10 (a)). However, [Ru(p-cymene)Cl2]2/AgSbF6-catalyzed reactions of N-(1H-pyrrolo [2,3-b]pyridin-1-yl)-benzamides with internal alkynes afford isoquinolone derivatives with remaining N-N bond untouched (Scheme 16) [71]. In this procedure, N-amino-7-azaindole is used as an efficient bidentate-directing group to promote the selective C–H bond activation.
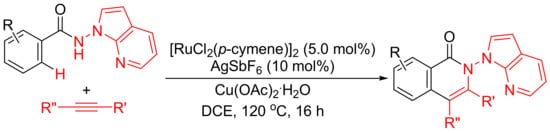
Scheme 16. Ruthenium-catalyzed isoquinolone formation by N-amino-7-azaindole-assisted C–H activation.
Instead of internal alkynes, the annulations of benzamides with other C2 synthons via C–H/N–H activation to construct isoquinolone ring have also been developed. For example, Glorius and coworkers developed a Rh(III)-catalyzed intermolecular [4 + 2] cyclocondensation of N-methoxybenzamides with α-halo and pseudohalo ketones to afford various 3-substituted N-methoxyisoquinolones in moderate to excellent yields (Scheme 17) [72]. The α-halo and pseudohalo ketones are utilized as oxidized alkyne equivalents to undergo the redox-neutral annulation, and through the formation of ortho-acylmethyl-benzamides as the intermediates. Recently, Ma and coworkers have also reported a Pd(OAc)2/L-alanine-catalyzed formation of 3-substituted N-(quinolin-8-yl)-isoquinolones with by N-(quinolin-8-yl)benzamides and α-bromo ketones in the presence of K2CO3 in t-AmOH [73].
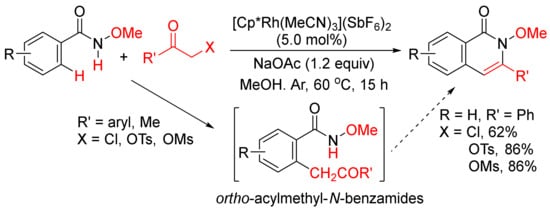
Scheme 17. 3-Substituted isoquinolone synthesis via Rh(III)-catalyzed intermolecular cyclocondensation of N-methoxybenzamides with α-halo and pseudohalo ketones.
With the use of same rhodium complex, 4-substituted isoquinolones can be obtained by a microwave-assisted annulation of N-methoxyamides with α-chloroaldehydes (Scheme 18) [74].
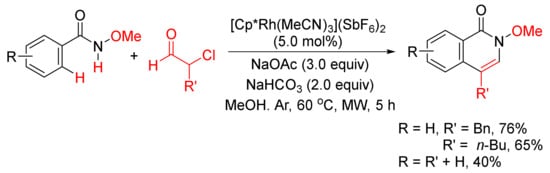
Scheme 18. 4-Substituted isoquinolone formation via microwave-assisted annulation of N-methoxyamides with α-chloroaldehydes.
Moreover, 3-Cyclohexylmethyl N-methoxyisoquinolone, which was published as only one example in literature [75], can be synthesized by the cyclocondensation of N-methoxyisoquinolone with the corresponding α-carbonyl sulfoxonium ylide catalyzed by rhodium in HFIP (Scheme 19). The detailed studies for the formation of 3-substituted isoquinolones through the reactions of N-methoxybenzamides and α-carbonyl sulfoxonium ylides catalyzed by Cp*Rh(MeCN)3(SbF6)2/Zn(OTf)2 in DCE were then reported by Li and coworkers [76].
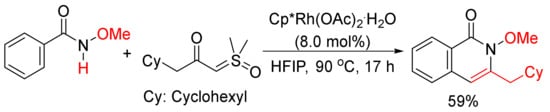
Scheme 19. Rhodium-catalyzed cyclocondensation of N-methoxyisoquinolone with α-carbonyl sulfoxonium ylide affording 3-substituted isoquinolone.
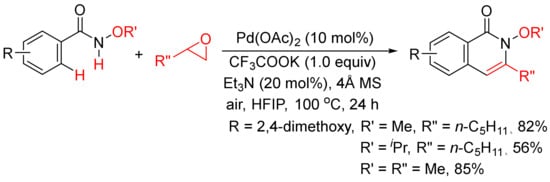
Very recently, epoxides as alkylating reagents and C2 synthon have been applied in Pd(OAc)2-catalyzed oxidative annulation of N-alkoxybenzamides to regioselectively synthesize 3-substituted isoquinolones in HFIP solvent (Scheme 20) [77]. The present methodology has been successfully employed in the total syntheses of rupreschstyril, siamine, and cassiarin A (
Figure 1).
).
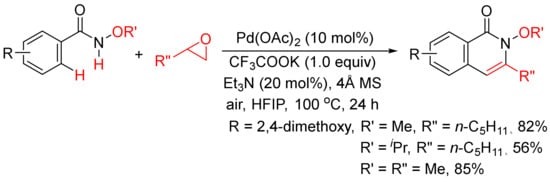
Scheme 20. Pd(OAc)2-catalyzed annulation of epoxides with benzamides.
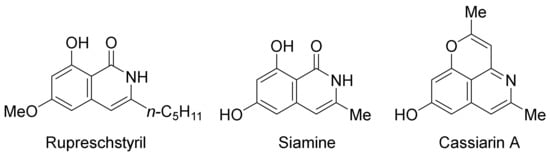
Figure 1. Synthetic applications in the total synthesis of natural products.
In addition, as depicted in Scheme 3, β-keto esters can be used as a C2 synthon in the formation of isoquinolones via cyclocondensation with ortho-halobenzamides catalyzed by CuI. However, in the presence of Pd(OOCCF3)2 and K2S2O8, the reactions of N-alkoxybenzamides with β-keto esters have also been developed for the synthesis of isoquinolones via selective C–H/N–H activation [78].
Moreover, as shown in Scheme 21, in the presence of [Cp*RhCl2]2, the regioselective formation of substituted isoquinolones through cyclocondensation of N-methoxybenz-amide [79] or primary benzamides [80] with diazo compounds has also been developed. In the case of N-methoxybenzamide used, C–H/N–H activation occurs selectively.
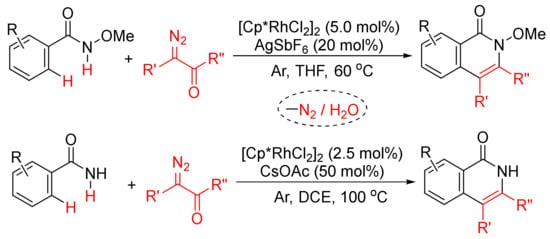
Scheme 21. Approach to isoquinolinones by rhodium-catalyzed annulation of benzamides with diazo compounds.
Similarly, [Cp*RhCl2]2/KOAc-catalyzed intermolecular cyclization between N-methoxybenzamide and 4-diazoisochroman-3-imines provides 3-amino-4-aryliso-quinolones via a selective N–O cleavage of 4-diazoisochroman-3-imines (Scheme 22) [81].
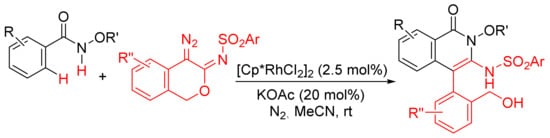
Scheme 22. Rh(III)-catalyzed synthesis of highly substituted isoquinolones from 4-diazoisochroman-3-imines and N-alkoxybenzamides.
The construction of isoquinolone ring can also be realized by the annulation of carbon–carbon double bonds with benzamides with selective C–H/N–H activation.
For example, with the use of Mn(OAc)3·2H2O as the oxidant and NaOPiv·H2O as the base in TFE, Thrimurtulu and coworkers developed a Co(acac)2-catalyzed isoquinolone synthesis by the intermolecular cyclization between N-(quinolin-8-yl)benzamides and substituted allenes at room temperature to give 3-substituted N-(quinolin-8-yl)isoquino- lones [82]. In addition, in the presence of Ag2CO3 and Cs2CO3, Co(OAc)2 can catalyze the similar cyclization in the same solvent giving isoquinolones [83].
The catalysts system of [Cp*Rh(MeCN)3][SbF6]2/Cu(OAc)2 in HFIP also shows highly catalytic activity in the cyclocondensation of N-methoxybenzamides with nitroalkenes for the selective synthesis of 4-substituted N-methoxyisoquinolones [84].
Scheme 20. Pd(OAc)2-catalyzed annulation of epoxides with benzamides.
3. Isoquinolone Syntheses via [5 + 1] Intermolecular Annulation between Ortho-(1-Alkynyl)Benzaldehyde and Primary Amines
Readily available ortho-(1-alkynyl)benzaldehydes have been well-applied in the synthesis of benzo-fused heterocycles [85,86,87,88,89,90,91] and carbocycles [92,93,94,95,96,97], which are also used as C5 synthon in the syntheses of isoquinolones by the intermolecular [5 + 1] cyclocondensation with primary amines.
As shown in Scheme 23, in the presence of SiO2, using an excess amount of CuBr·SMe2, the aerobic cyclocondensation of ortho-(1-alkynyl)benzaldehydes with benzyl amines or primary aliphatic amines affords 4-bromoisoquinolones [98]. Cu(OAc)2-catalyzed cyclocondensation of ortho-(1-alkynyl)benzaldehydes with aniline gives 2,3-diphenylisoquinolone in 63% yield [99].
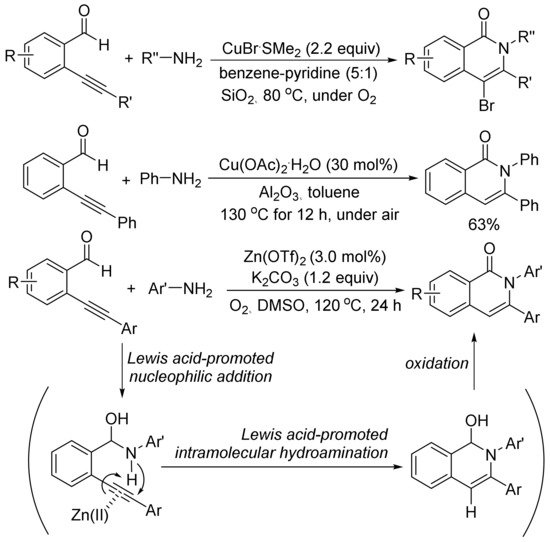
Scheme 23. Isoquinolone formation via [5 + 1] cyclocondensation of ortho-(1-alkynyl)-benzaldehydes with amines.
Zn(OTf)2 also shows catalytic activity for the aerobic cyclocondensation of ortho-(1-alkynyl)benzaldehydes with arylamines to give N-arylated isoquinolones [100]. The proposed mechanism involves two well-known steps of nucleophilic addition of arylamines to aldehyde, and intramolecular hydroamination followed by oxidation reaction affording isoquinolone. Zn(OTf)2 plays an important role to promote both nucleophilic addition of arylamines and intramolecular hydroamination of carbon–carbon triple bond.
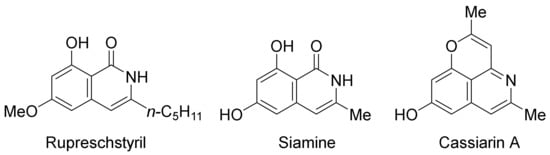
Figure 1. Synthetic applications in the total synthesis of natural products.
4. Isoquinolone Syntheses via Other Intermolecular Annulations
A Ni(cod)2/phosphine-catalyzed denitrogenative alkyne insertion reaction of 1,2,3-benzotriazin-4(3H)-ones provides an alternatively efficient approach to substituted isoquinolones (Scheme 24) [101]. A wide-range isoquinolones can be obtained in high yields with the use of internal/terminal alkynes, including borylalkynes. This transformation can also be realized under visible-light irradiation with the assistance of a photocatalyst at room temperature [102].
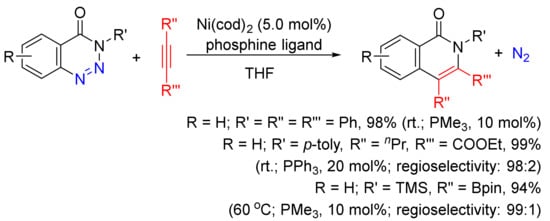
Scheme 24. Isoquinolone formation via nickel-catalyzed denitrogenative alkyne insertion of 1,2,3-benzotriazin-4(3H)-ones.
In addition, the similar catalyst system can also catalyze a decarbonylative addition of phthalimides to internal alkynes to afford highly substituted isoquinolones (Scheme 25) [103]. The similar transformation can take place using CoI2/phosphine/Ag2CO3 [104], and Rh(PPh3)2(CO)Cl as the catalyst systems [105].
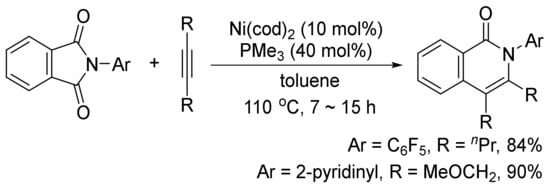
Scheme 25. Nickel-catalyzed decarbonylative addition of phthalimides to alkynes.
The isoquinolone skeleton can also be constructed by a Pd(OAc)2-catalyzed carbonylation/decarboxylation of diethyl(2-iodoaryl)malonates with imidoyl chlorides and CO in THF (Scheme 26) [106].
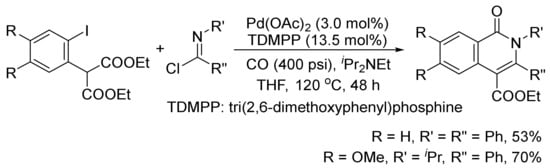
Scheme 26. Palladium-catalyzed construction of isoquinolone ring by carbonylation/decarboxylation reaction.
The annulation reaction between benzocyclobutenols and isocyanates catalyzed by [RhOH(cod)]2 can also produce isoquinolone derivatives with excellent regioselectivity and functional-group tolerance (Scheme 27) [107]. The reactions give the first examples of formal amide fragment insertion into an aryl C–C(sp3) bond.
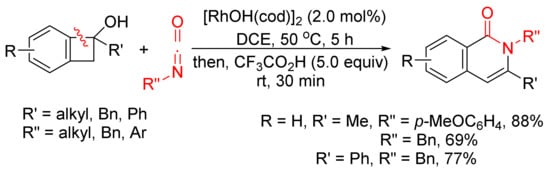
Scheme 27. Rhodium(I)-catalyzed cyclocondensation of benzocyclobutenols and isocyanates affording 3-substituted isoquinolone.
In addition, as depicted in Scheme 3, β-keto esters can be used as a C2 synthon in the formation of isoquinolones via cyclocondensation with ortho-halobenzamides catalyzed by CuI. However, in the presence of Pd(OOCCF3)2 and K2S2O8, the reactions of N-alkoxybenzamides with β-keto esters have also been developed for the synthesis of isoquinolones via selective C–H/N–H activation [78].
5. Isoquinolone Syntheses via Intramolecular Annulation Reactions
Isoquinolone syntheses via intramolecular annulation can be achieved starting from a variety of substrates. For example, the intramolecular cyclization of phosphorylated aromatic carboxamides promoted by base [108], palladium-catalyzed intramolecular cyclization of ortho-ethynylbenzamides [109], ortho-iodo-N-allenylbenzamides [110], palladium-catalyzed, or base-promoted intramolecular cyclization of ortho-allenyl-N-substituted benzamides generated in situ [111].
When ortho-(2-substituted ethynyl)benzonitriles and NaOMe were refluxed in methanol for 16 h, both isoindolones and isoquinolones formed in different ratios, depending on the nature of substituents. Substrates bearing the phenyl and thienyl group and stabilizing the α-anion favor the 5-exo-pathway to give isoindolones, whereas pyridinyl and pyrazinyl groups can also stabilize the α-anion, but the formation of a more stable intermediate by coordination of sodium with nitrogen atom leads to the formation of isoquinolones via 6-endo cyclization (Scheme 28) [112].
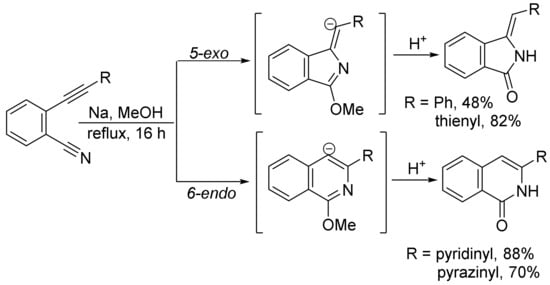
Scheme 28. Formation of isoindolones and isoquinolones.
In addition, in the presence of platinum complexes, the intramolecular cyclization of ortho-alkynylbenzonitriles in alcohols produces 3-substituted isoquinolones [113].
Treatment of ortho-iodobenzoyl azides with terminal alkynes in the presence of Pd/C, PPh3, and CuI in EtOH at 80 °C under N2 afforded 3-substituted isoquinolones as the major products [114]. The formation of isoquinolones is proposed involving in situ generation of ortho-alkynyl benzoyl azides via Pd/C-mediated Sonogashira coupling followed by intramolecular acetylenic Schmidt reaction with denitrogenation (Scheme 29). Aliphatic terminal alkynes show high chemoselectivity to give the corresponding isoquinolones in good to high yields, whereas the use of aryl alkynes affords the desired products in low yield along with other side products.
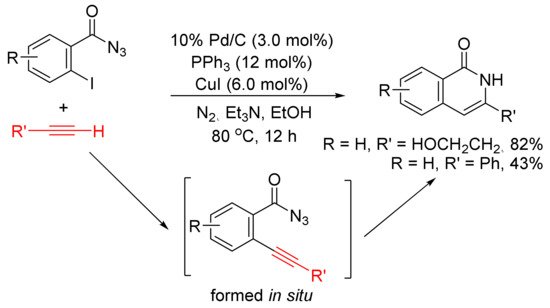
Scheme 29. Isoquinolone formation via intramolecular cyclization of ortho-alkynylbenzoyl azides.
In the presence of H2O, Cu(OAc)2.H2O-catalyzed tandem cyclization reaction of 2-(1-alkynyl)arylaldimines produces highly substituted isoquinolone derivatives (Scheme 30) [99].
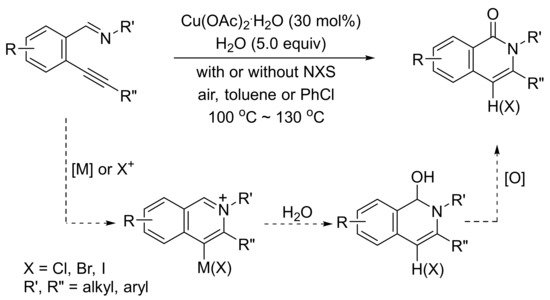
Scheme 30. Isoquinolone formation by copper(II)-catalyzed tandem reactions of 2-(1-alkynyl)benzaldimines with water.
Moreover, 4-substituted isoquinolones can be prepared by a DBU-promoted isomerization of 4-(diarylmethylidene)-3,4-dihydroisoquinolinones, which are generated via the palladium-catalyzed tandem Heck-Suzuki coupling reactions of ortho-iodo-N-(prop-2-ynyl)-benzamides with arylboronic acids under mild conditions (Scheme 31) [115].
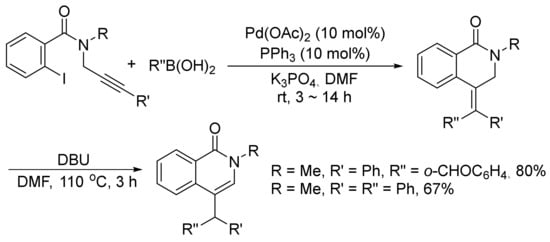
Scheme 31. Base-promoted isoquinolone synthesis via isomerization of 4-methylidene-3,4-dihydroisoquinolinones.
As shown in Scheme 8, the [4 + 2] cyclocondensation of N-alkoxybenzamides with vinyl acetate gives 3,4-unsubstituted N–H isoquinolones, and vinyl acetate is a good acetylene equivalent. The other approach to 3,4-unsubstituted N–H isoquinolones is designed from the acid-promoted intramolecular cyclization of ortho-vinyl ether benzamides, which are pre-prepared by Suzuki cross-coupling reactions of the corresponding pinacol borate esters with ortho-bromobenzamides (Scheme 32 (a)) [116]. Using trifluoroacetic acid (TFA) as a solvent, the intramolecular cyclization occurs to form isoquinolone ring with t-butyl group removed under the strong acidic conditions and microwave irradiation. A similar procedure was reported under mild conditions promoted by HCl in dioxane to give N-substituted 3,4-unsubstituted isoquinolones (Scheme 32 (b)) [117].
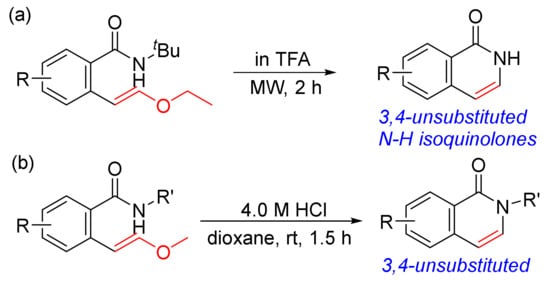
Scheme 32. 3,4-Unsubstituted isoquinolones via acid-promoted intramolecular cyclization of ortho-vinyl ether benzamides.
Moreover, as shown in Scheme 21, in the presence of [Cp*RhCl2]2, the regioselective formation of substituted isoquinolones through cyclocondensation of N-methoxybenz-amide [79] or primary benzamides [80] with diazo compounds has also been developed. In the case of N-methoxybenzamide used, C–H/N–H activation occurs selectively.
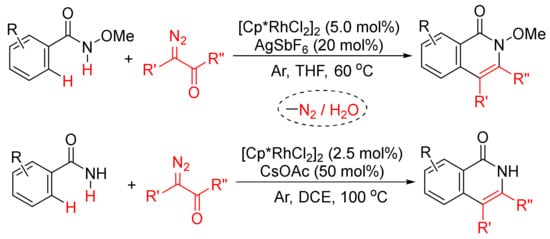
Scheme 21. Approach to isoquinolinones by rhodium-catalyzed annulation of benzamides with diazo compounds.
Similarly, [Cp*RhCl2]2/KOAc-catalyzed intermolecular cyclization between N-methoxybenzamide and 4-diazoisochroman-3-imines provides 3-amino-4-aryliso-quinolones via a selective N–O cleavage of 4-diazoisochroman-3-imines (Scheme 22) [81].
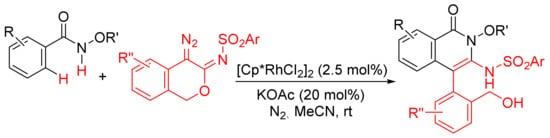
Scheme 22. Rh(III)-catalyzed synthesis of highly substituted isoquinolones from 4-diazoisochroman-3-imines and N-alkoxybenzamides.
The construction of isoquinolone ring can also be realized by the annulation of carbon–carbon double bonds with benzamides with selective C–H/N–H activation.
For example, with the use of Mn(OAc)3·2H2O as the oxidant and NaOPiv·H2O as the base in TFE, Thrimurtulu and coworkers developed a Co(acac)2-catalyzed isoquinolone synthesis by the intermolecular cyclization between N-(quinolin-8-yl)benzamides and substituted allenes at room temperature to give 3-substituted N-(quinolin-8-yl)isoquino- lones [82]. In addition, in the presence of Ag2CO3 and Cs2CO3, Co(OAc)2 can catalyze the similar cyclization in the same solvent giving isoquinolones [83].
The catalysts system of [Cp*Rh(MeCN)3][SbF6]2/Cu(OAc)2 in HFIP also shows highly catalytic activity in the cyclocondensation of N-methoxybenzamides with nitroalkenes for the selective synthesis of 4-substituted N-methoxyisoquinolones [84].
3. Isoquinolone Syntheses via [5 + 1] Intermolecular Annulation between Ortho-(1-Alkynyl)Benzaldehyde and Primary Amines
Readily available ortho-(1-alkynyl)benzaldehydes have been well-applied in the synthesis of benzo-fused heterocycles [85,86,87,88,89,90,91] and carbocycles [92,93,94,95,96,97], which are also used as C5 synthon in the syntheses of isoquinolones by the intermolecular [5 + 1] cyclocondensation with primary amines.
As shown in Scheme 23, in the presence of SiO2, using an excess amount of CuBr·SMe2, the aerobic cyclocondensation of ortho-(1-alkynyl)benzaldehydes with benzyl amines or primary aliphatic amines affords 4-bromoisoquinolones [98]. Cu(OAc)2-catalyzed cyclocondensation of ortho-(1-alkynyl)benzaldehydes with aniline gives 2,3-diphenylisoquinolone in 63% yield [99].
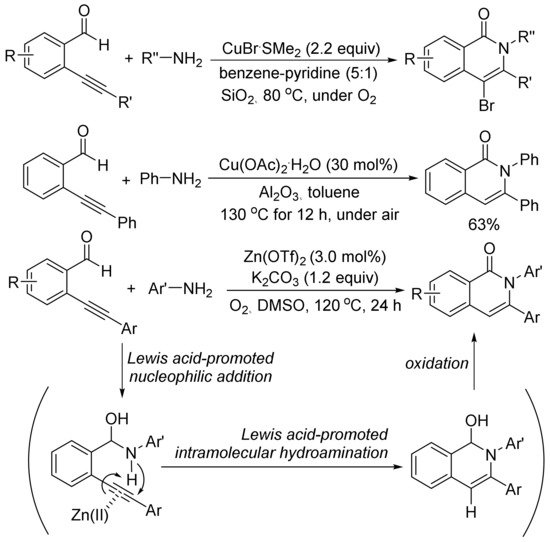
Scheme 23. Isoquinolone formation via [5 + 1] cyclocondensation of ortho-(1-alkynyl)-benzaldehydes with amines.
Zn(OTf)2 also shows catalytic activity for the aerobic cyclocondensation of ortho-(1-alkynyl)benzaldehydes with arylamines to give N-arylated isoquinolones [100]. The proposed mechanism involves two well-known steps of nucleophilic addition of arylamines to aldehyde, and intramolecular hydroamination followed by oxidation reaction affording isoquinolone. Zn(OTf)2 plays an important role to promote both nucleophilic addition of arylamines and intramolecular hydroamination of carbon–carbon triple bond.
4. Isoquinolone Syntheses via Other Intermolecular Annulations
A Ni(cod)2/phosphine-catalyzed denitrogenative alkyne insertion reaction of 1,2,3-benzotriazin-4(3H)-ones provides an alternatively efficient approach to substituted isoquinolones (Scheme 24) [101]. A wide-range isoquinolones can be obtained in high yields with the use of internal/terminal alkynes, including borylalkynes. This transformation can also be realized under visible-light irradiation with the assistance of a photocatalyst at room temperature [102].
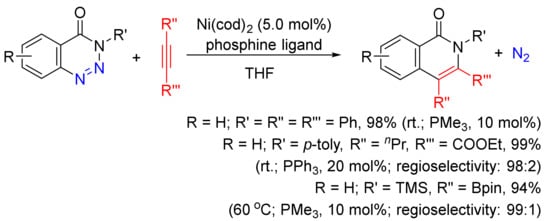
Scheme 24. Isoquinolone formation via nickel-catalyzed denitrogenative alkyne insertion of 1,2,3-benzotriazin-4(3H)-ones.
In addition, the similar catalyst system can also catalyze a decarbonylative addition of phthalimides to internal alkynes to afford highly substituted isoquinolones (Scheme 25) [103]. The similar transformation can take place using CoI2/phosphine/Ag2CO3 [104], and Rh(PPh3)2(CO)Cl as the catalyst systems [105].
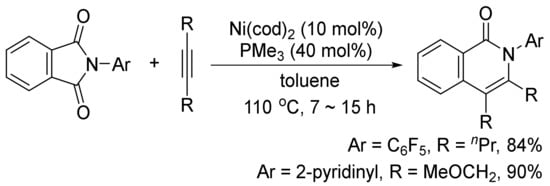
Scheme 25. Nickel-catalyzed decarbonylative addition of phthalimides to alkynes.
The isoquinolone skeleton can also be constructed by a Pd(OAc)2-catalyzed carbonylation/decarboxylation of diethyl(2-iodoaryl)malonates with imidoyl chlorides and CO in THF (Scheme 26) [106].
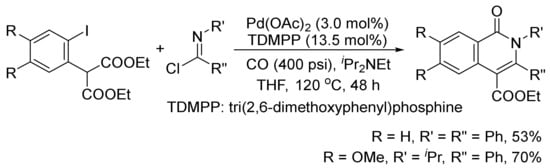
Scheme 26. Palladium-catalyzed construction of isoquinolone ring by carbonylation/decarboxylation reaction.
The annulation reaction between benzocyclobutenols and isocyanates catalyzed by [RhOH(cod)]2 can also produce isoquinolone derivatives with excellent regioselectivity and functional-group tolerance (Scheme 27) [107]. The reactions give the first examples of formal amide fragment insertion into an aryl C–C(sp3) bond.
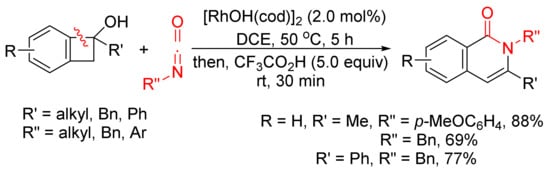
Scheme 27. Rhodium(I)-catalyzed cyclocondensation of benzocyclobutenols and isocyanates affording 3-substituted isoquinolone.
5. Isoquinolone Syntheses via Intramolecular Annulation Reactions
Isoquinolone syntheses via intramolecular annulation can be achieved starting from a variety of substrates. For example, the intramolecular cyclization of phosphorylated aromatic carboxamides promoted by base [108], palladium-catalyzed intramolecular cyclization of ortho-ethynylbenzamides [109], ortho-iodo-N-allenylbenzamides [110], palladium-catalyzed, or base-promoted intramolecular cyclization of ortho-allenyl-N-substituted benzamides generated in situ [111].
When ortho-(2-substituted ethynyl)benzonitriles and NaOMe were refluxed in methanol for 16 h, both isoindolones and isoquinolones formed in different ratios, depending on the nature of substituents. Substrates bearing the phenyl and thienyl group and stabilizing the α-anion favor the 5-exo-pathway to give isoindolones, whereas pyridinyl and pyrazinyl groups can also stabilize the α-anion, but the formation of a more stable intermediate by coordination of sodium with nitrogen atom leads to the formation of isoquinolones via 6-endo cyclization (Scheme 28) [112].
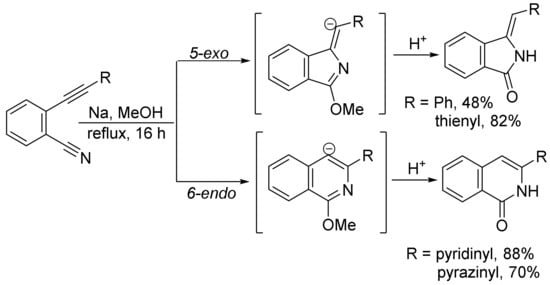
Scheme 28. Formation of isoindolones and isoquinolones.
In addition, in the presence of platinum complexes, the intramolecular cyclization of ortho-alkynylbenzonitriles in alcohols produces 3-substituted isoquinolones [113].
Treatment of ortho-iodobenzoyl azides with terminal alkynes in the presence of Pd/C, PPh3, and CuI in EtOH at 80 °C under N2 afforded 3-substituted isoquinolones as the major products [114]. The formation of isoquinolones is proposed involving in situ generation of ortho-alkynyl benzoyl azides via Pd/C-mediated Sonogashira coupling followed by intramolecular acetylenic Schmidt reaction with denitrogenation (Scheme 29). Aliphatic terminal alkynes show high chemoselectivity to give the corresponding isoquinolones in good to high yields, whereas the use of aryl alkynes affords the desired products in low yield along with other side products.
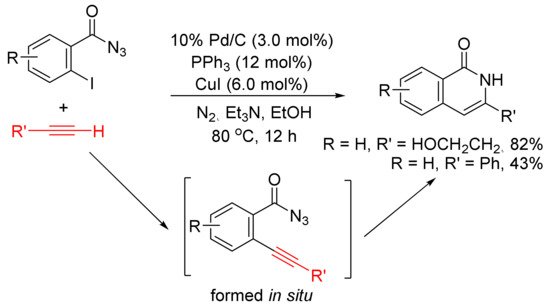
Scheme 29. Isoquinolone formation via intramolecular cyclization of ortho-alkynylbenzoyl azides.
In the presence of H2O, Cu(OAc)2.H2O-catalyzed tandem cyclization reaction of 2-(1-alkynyl)arylaldimines produces highly substituted isoquinolone derivatives (Scheme 30) [99].
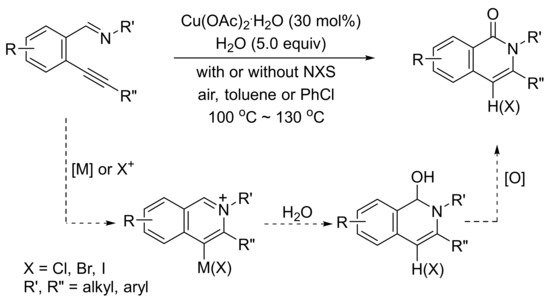
Scheme 30. Isoquinolone formation by copper(II)-catalyzed tandem reactions of 2-(1-alkynyl)benzaldimines with water.
Moreover, 4-substituted isoquinolones can be prepared by a DBU-promoted isomerization of 4-(diarylmethylidene)-3,4-dihydroisoquinolinones, which are generated via the palladium-catalyzed tandem Heck-Suzuki coupling reactions of ortho-iodo-N-(prop-2-ynyl)-benzamides with arylboronic acids under mild conditions (Scheme 31) [115].
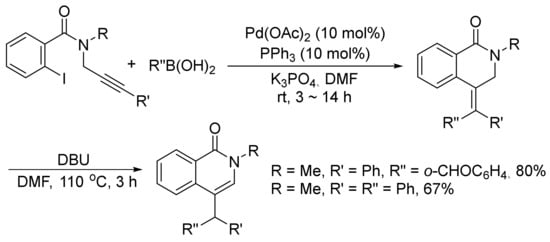
Scheme 31. Base-promoted isoquinolone synthesis via isomerization of 4-methylidene-3,4-dihydroisoquinolinones.
As shown in Scheme 8, the [4 + 2] cyclocondensation of N-alkoxybenzamides with vinyl acetate gives 3,4-unsubstituted N–H isoquinolones, and vinyl acetate is a good acetylene equivalent. The other approach to 3,4-unsubstituted N–H isoquinolones is designed from the acid-promoted intramolecular cyclization of ortho-vinyl ether benzamides, which are pre-prepared by Suzuki cross-coupling reactions of the corresponding pinacol borate esters with ortho-bromobenzamides (Scheme 32 (a)) [116]. Using trifluoroacetic acid (TFA) as a solvent, the intramolecular cyclization occurs to form isoquinolone ring with t-butyl group removed under the strong acidic conditions and microwave irradiation. A similar procedure was reported under mild conditions promoted by HCl in dioxane to give N-substituted 3,4-unsubstituted isoquinolones (Scheme 32 (b)) [117].
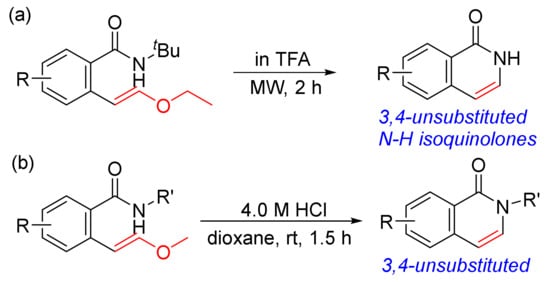
Scheme 32. 3,4-Unsubstituted isoquinolones via acid-promoted intramolecular cyclization of ortho-vinyl ether benzamides.
6. Polycyclic-Fused Isoquinolone Formation via Alkyne Annulation
Annulation protocols under different conditions have been extensively studied for one-pot and efficient approach to polycyclic-fused isoquinolones [118,119,120,121]. Transition metal-catalyzed procedures focus on the use of [Cp*RhCl2]2, [RuCl2(p-cymene)]2, and CuCl catalysts, through the intermolecular annulation of alkynes with benzamides and intramolecular annulation of alkyne-tethered benzamides, which are reviewed in this section.
In 2010, Li and coworker reported a [Cp*RhCl2]2-catalyzed double oxidative cyclocoupling of primary benzamides with two molecules of alkynes affording tetracyclic isoquinolones resulting from the formation of two C-N bonds and two C-C bonds simultaneously with the use of Ag2CO3 as an oxidant (Scheme 33) [52], which is similar to the use of Cu(OAc)2.2H2O as the oxidant as shown in Scheme 13. In the presence of CsOAc, the same catalyst also catalyzes the cyclization of N-(pivaloyloxy)benzamide with cyclohexadienone-containing 1,6-enynes, providing one-pot synthesis of tetracyclic isoquinolones under mild conditions (Scheme 34) [122].
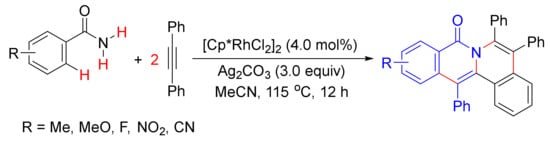
Scheme 33. Tetracyclic isoquinolone formation by Rh(III)-catalyzed double oxidative cyclocoupling between primary benzamides and alkynes.
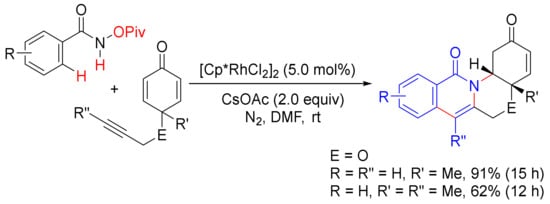
Scheme 34. Tetracyclic isoquinolone formation by Rh(III)-catalyzed cyclization of N-(pivaloyloxy)benzamide with 1,6-enynes.
In the presence of additive and/or oxidant, [Cp*RhCl2]2-catalyzed formation of other polycyclic-fused isoquinolones is shown in
6. Polycyclic-Fused Isoquinolone Formation via Alkyne Annulation
Annulation protocols under different conditions have been extensively studied for one-pot and efficient approach to polycyclic-fused isoquinolones [118,119,120,121]. Transition metal-catalyzed procedures focus on the use of [Cp*RhCl2]2, [RuCl2(p-cymene)]2, and CuCl catalysts, through the intermolecular annulation of alkynes with benzamides and intramolecular annulation of alkyne-tethered benzamides, which are reviewed in this section.
In 2010, Li and coworker reported a [Cp*RhCl2]2-catalyzed double oxidative cyclocoupling of primary benzamides with two molecules of alkynes affording tetracyclic isoquinolones resulting from the formation of two C-N bonds and two C-C bonds simultaneously with the use of Ag2CO3 as an oxidant (Scheme 33) [52], which is similar to the use of Cu(OAc)2.2H2O as the oxidant as shown in Scheme 13. In the presence of CsOAc, the same catalyst also catalyzes the cyclization of N-(pivaloyloxy)benzamide with cyclohexadienone-containing 1,6-enynes, providing one-pot synthesis of tetracyclic isoquinolones under mild conditions (Scheme 34) [122].
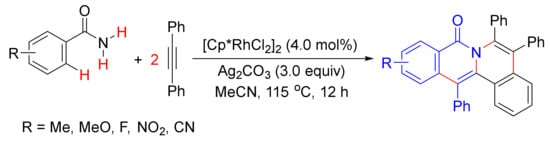
Scheme 33. Tetracyclic isoquinolone formation by Rh(III)-catalyzed double oxidative cyclocoupling between primary benzamides and alkynes.
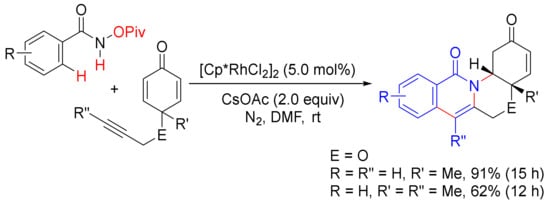
Scheme 34. Tetracyclic isoquinolone formation by Rh(III)-catalyzed cyclization of N-(pivaloyloxy)benzamide with 1,6-enynes.
In the presence of additive and/or oxidant, [Cp*RhCl2]2-catalyzed formation of other polycyclic-fused isoquinolones is shown in
Figure 2. Furthermore, 2,3-Dihydropyrrolo [1,2-b] isoquinolin-5(1H)-ones (n = 1) [123], and isoindolo[2,1-b]isoquinolin-5(7H)-ones (n = 1) [124] can be prepared by intramolecular annulations of alkyne tethered benzamides, and the latter approach is used in the synthesis of nitrogen-containing natural products such as rosettacin and oxypalmatime. The intermolecular cascade annulation between N-pivaloyloxy benzamides and ortho-alkynylbenzaldehydes can give 7-hydroxyisoin-dolo[2,1-b]-isoquinolin-5(7H)-ones and also afford the convenient intermediates for access to rosettacin [125,126]. In addition, the intermolecular cyclization of N-pivaloyloxy benzamides with conjugated enynones affords pyrrolo[1,2-b]isoquinolin-5(3H)-ones [127]. All the formed polycyclic isoquinolones are not easily available to be synthesized by multi-step traditional organic transformation.
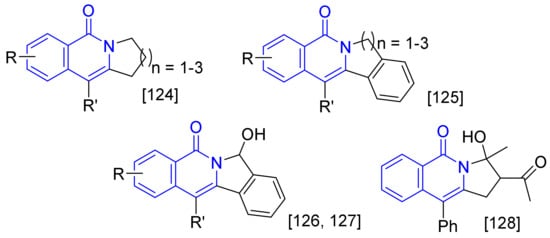
. Furthermore, 2,3-Dihydropyrrolo [1,2-b] isoquinolin-5(1H)-ones (n = 1) [123], and isoindolo[2,1-b]isoquinolin-5(7H)-ones (n = 1) [124] can be prepared by intramolecular annulations of alkyne tethered benzamides, and the latter approach is used in the synthesis of nitrogen-containing natural products such as rosettacin and oxypalmatime. The intermolecular cascade annulation between N-pivaloyloxy benzamides and ortho-alkynylbenzaldehydes can give 7-hydroxyisoin-dolo[2,1-b]-isoquinolin-5(7H)-ones and also afford the convenient intermediates for access to rosettacin [125,126]. In addition, the intermolecular cyclization of N-pivaloyloxy benzamides with conjugated enynones affords pyrrolo[1,2-b]isoquinolin-5(3H)-ones [127]. All the formed polycyclic isoquinolones are not easily available to be synthesized by multi-step traditional organic transformation.
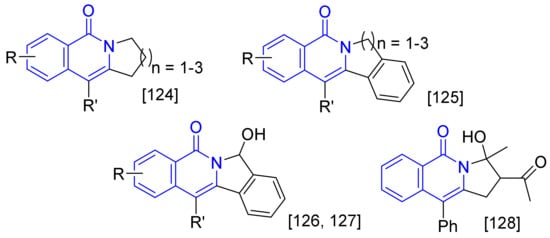
Figure 2. Polycyclic fused isoquinolones.
As described in Scheme 7, [Cp*RhCl2]2/CsOAc-catalyzed intramolecular reaction of alkyne-tethered N-alkoxybenzamides leads to 3-hydroxyalkyl-substituted N–H isoquinolones by selective C–H/N–O activation [31]. However, CuCl-catalyzed aerobic oxidative radical cascade annulations of the similar substrates give isoxazolidine/1,2-oxazinane-fused isoquinolones, and N–O bond is untouched (Scheme 35) [128]. This transformation features air as the environment-friendly oxidant and has the merits of a cheap catalyst, broad substrates scope, high atom economy, and simple operation.
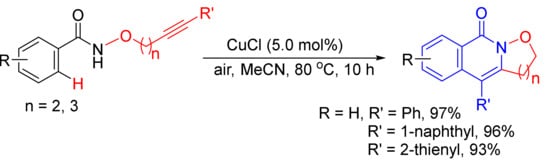
Scheme 35. CuCl-catalyzed synthesis of isoxazolidine-fused isoquinolones.
Very recently, the formation of isoxazolidine-fused isoquinolones starting from alkyne-tethered N-alkoxybenzamides has been reported by electrochemical-oxidation-induced [129] and PhI(OAc)2-mediated [130] intramolecular annulations.
Figure 2. Polycyclic fused isoquinolones.
As described in Scheme 7, [Cp*RhCl2]2/CsOAc-catalyzed intramolecular reaction of alkyne-tethered N-alkoxybenzamides leads to 3-hydroxyalkyl-substituted N–H isoquinolones by selective C–H/N–O activation [31]. However, CuCl-catalyzed aerobic oxidative radical cascade annulations of the similar substrates give isoxazolidine/1,2-oxazinane-fused isoquinolones, and N–O bond is untouched (Scheme 35) [128]. This transformation features air as the environment-friendly oxidant and has the merits of a cheap catalyst, broad substrates scope, high atom economy, and simple operation.
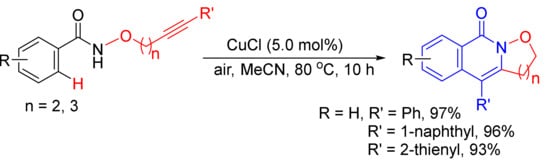
Scheme 35. CuCl-catalyzed synthesis of isoxazolidine-fused isoquinolones.
Very recently, the formation of isoxazolidine-fused isoquinolones starting from alkyne-tethered N-alkoxybenzamides has been reported by electrochemical-oxidation-induced [129] and PhI(OAc)2-mediated [130] intramolecular annulations.
[Ru(p-cymene)Cl2]2 not only shows the catalytic activity for the construction of isoquinolone ring as described-above but also has been employed as catalyst in the formation of polycyclic-fused isoquinolones, such as tetracyclic isoquinolones [131] and spiro-fused-isoquinolones [132] (Figure 3).
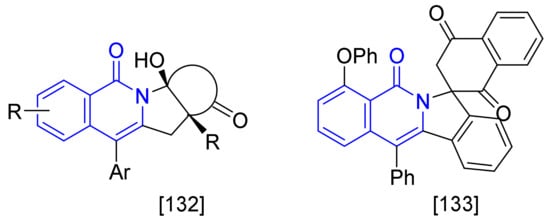
Figure 3. Ruthenium(II)-catalyzed syntheses of polycyclic fused isoquinolones.
).
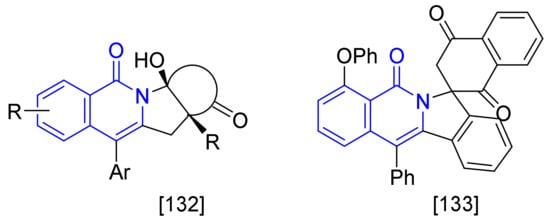
Figure 3. Ruthenium(II)-catalyzed syntheses of polycyclic fused isoquinolones.