Pancreatic ductal adenocarcinoma (PDAC) is currently the third leading cause of cancer-related mortality. About 90% of PDAC cases are diagnosed in patients older than 55 years, and with increased longevity in the general population PDAC burden is expected to rise. Still, the survival is abysmal, with a 5-year survival rate of 8.2%.
- pancreatic cancer
- microbiome
- mycobiome
- inflammation
- immunosuppression
- tumor initiation
- tumor progression
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is currently the third leading cause of cancer-related mortality. About 90% of PDAC cases are diagnosed in patients older than 55 years, and with increased longevity in the general population PDAC burden is expected to rise. Still, the survival is abysmal, with a 5-year survival rate of 8.2% [1].
Surgical resection is currently considered to be the only curative treatment. However, only 15% of patients present with a resectable disease at diagnosis [1]. Moreover, patients undergoing resection and adjuvant chemotherapy have a limited prognosis with a median overall survival between 28 and 54 months [2][3][4][2,3,4].
PDAC occurs mostly sporadically. Risk factors include smoking, heavy alcohol intake, history of chronic pancreatitis (CP), overweight, and diabetes. This microbial community presents a wide inter-individual variability, depending on host-specific factors, such as age, gender, genotype, and bile acids production [5][7]. Of note, it has been shown that the pancreas can also shape the flora by specific antimicrobial peptide secretion [6][8].
The intestinal micro- and mycobiome have recently gained increasing interest in the field of PDAC with studies suggesting a tumorigenic relevance of both bacterial and fungal dysbiosis. This review aims to give an overview of the alteration patterns of bacterial and fungal flora associated with PDAC and to highlight possible molecular pathways linking bacterial and fungal dysbiosis with pancreatic carcinogenesis.
2. Microbiome Alterations and PDAC
The human intestine bears more than 5.000 different bacterial species (1014microorganisms), which are fundamental for regulating the balance between health and disease [7][9]. Microbial dysbiosis and disrupted epithelial barriers can promote bacterial translocation favoring neoplastic transformation. The microbiome’s oncogenesis contribution has emerged in different malignancies of the gastrointestinal tract, such as esophageal, gastric and colorectal carcinoma [7][8][9,10]. However, only few studies associate the gut flora with tumor development of non-gastrointestinal tract tissues.
Alterations of the oral microbiota have been linked to PDAC in different studies. In periodontal disease, pathogenetic oral flora and tooth loss have been described as independent risk factors for PDAC development [9][10][11][12,13,14]. [12][15], and correspondent plasma antibodies were elevated in PDAC subjects [13][16]. Of note in this context,P. gingivalishas been demonstrated to be able to survive both inside human and murine pancreatic cancer cells in vitro, especially under hypoxic conditions, which is a typical trait of PDAC [14][17].
On the contrary, some other bacterial taxa likeVeillonellaspp. andNeisseria elongatahave been found to be negatively associated with PDAC, thus representing a possible protective factor against this type of malignancy [12][15][16][15,18,19].
Curiously, contrasting results come from the analysis ofStreptococcus,Fusobacterium nucleatumandLeptorichia. While the carrier-status related to these taxa was described by some groups to be associated with a decreased risk of developing PDAC [12][16][17][15,19,20], others found a positive correlation [15][18][18,21]. Interestingly, one study showed higher serum and salivary antibodies againstF. nucleatumin patients with high-grade dysplasia intraductal papillary mucinous neoplasms (IPMN) or IPMN with associated invasive cancer compared to patients bearing a low-risk IPMN [19][22]. However, other studies could not confirm these differences [20][23].
Results on the correlation betweenHelicobacterspp. and PDAC are inconsistent. Serological analysis showed in some studies a positive correlation betweenH. pyloriand PDAC [10][21][22][23][13,24,25,26], however, CagA-positive strains ofH. pylorishowed no significant association with pancreatic cancer [13][24][25][26][27][16,27,28,29,30]. In this sense, an increased risk of developing PDAC in the presence of CagA-negativeHelicobacterstrains could be conceivable.
While some groups did not find any correspondent DNA in the pancreatic tissue or juice [28][31], others isolated DNA in tumor tissue of PDAC patients but not in healthy controls [29][32]. Interestingly, in this study, the DNA of entericHelicobacterspecies andH. pyloriwas never present in both the pancreatic and the gastroduodenal tissue. This suggests that migration from the gut into the pancreas seems to be unlikely.
The analysis of pancreatic samples and fluids from patients with PDAC compared to samples from patients with a healthy organ or with benign pathologies provided evidence that the pancreas is not a sterile organ [29][30][31][32][33][34][35][36][37][38][39][40][41][32,33,34,35,36,37,38,39,40,41,42,43,44] (Table 1). In particular, the analysis of pancreatic cystic fluid revealed a specific bacterial ecosystem which may reflect the microbiota harbored within the pancreas [36][39].
| Reference | Detection Method(s) |
Type and Size of Pancreas Sample | Contamination Evaluation | Findings | Conclusion |
|---|---|---|---|---|---|
| Nilsson et al. [29], 2006, Sweden | 16S rRNA PCR | 40 PDAC tissue 14 NET 1 tissue 8 MEN1 2 tissue 5 CP 3 tissue 10 benign diseases 7 normal tissue |
PCR contamination prevention | 75% PDAC, 60% CP positive for Helicobacter DNA. Benign and healthy negative |
Possible role of Helicobacter in CP and PDAC development |
| Mitsuhashi et al. [30], Japan, 2015 | 16S rRNA PCR qPCR | 302 PDAC tissue 25 normal tissue |
Not available | 8.8% PDAC positive for Fusobacterium sp. | Fusobacterium correlates with worse PDAC prognosis |
| Geller et al. [34], USA, 2017 |
16S rRNA PCR | 20 normal tissue 113 PDAC tissue |
Negative control sample | Enterobacteriaceae and Pseudomonas prevalent in PDAC | Bacteria are a component of the PDAC tumor microenvironment |
| Rogers et al. [35], USA, 2017 |
16S rRNA PCR qPCR | 50 PDAC tissue | PCR contamination prevention | PDAC enriched with Klebsiella and Acinetobacter | Bacteria are a component of the PDAC tumor microenvironment |
| Li et al. [36], The Nedetherlands, 2017 | 16S rRNA PCR NGS 4 |
69 pancreatic cystic fluid | Extrapancreatic control sample (duodenum), Bioinformtaic tools |
Bacteroides spp., Enterobacteriaceae, Acidaminococcus spp. prevalent in cystic fluid | Pancreatic cysts harbor a specific bacterial ecosystem with possible role in the neoplastic process |
| Maekawa et al. [37], Japan, 2018 | 16S rRNA PCR | 5 PDAC tissue 20 PDAC juice |
Not available | PDAC juice and tissue samples mostly positive for Enterococcus faecalis | Possible role of E. faecalis in CP and PDAC development |
| Pushalkar et al. [38], USA, 2018 | 16S rRNA PCR qPCR FISH 5 |
12 PDAC tissue | PCR contamination prevention | Proteobacteria, Bacteroidetes and Firmicutes prevalent in PDAC | Bacteria are a component of the PDAC tumor microenvironment |
| Riquelme et al. [40], USA, 2019 |
16S rRNA PCR rRNA FISH |
68 PDAC tissue | PCR contamination prevention, bioinformatic tools | Proteobacteria Actinobacteria and Bacillus clausii correlate with PDAC long-term survivors | Microbiome diversity determines the survival of PDAC patients |
| Del Castillo et al. [41], USA, 2019 |
16S rRNA PCR | 51 PDAC tissue 18 CP tissue 8 other (bile duct, small bowel diseases) 34 normal tissue |
Physical specimen manipulation | Proteobacteria, Firmicutes Bacteroides, Fusobacteria and Actinobacteria prevalent in PDAC. Lactobacillus in non-cancer subjects | Different microbiome composition between PDAC and normal pancreas |
| Aykut et al. [31], USA, 2019 |
18S rRNA PCR FISH 18S ITS 6 sequencing |
13 PDAC tissue | Negative control sample | Ascomycota and Basidiomycota phyla and Malassezia genus prevalent in PDAC | Fungi are a component of the PDAC tumor microenvironment |
| Chakladar et al. [32], USA, 2020 |
16S rRNA PCR | 187 PDAC tissue | Bioinformatic tools | Proteobacteria prevalent in PDAC. Pseudomonadales Acidovorax ebreus C. freundii. S. sonnei related to worse prognosis A. baumannii and M. hypopneumoniae correlate with smoke-related PDAC A. ebreus, C. baumannii and G. kaustophilus and E. coli prevalent in male PDAC |
Corroboration of previous results. 13 microbes correlated to the dysregulation of gene signatures related to oncogenic methylation, cancer progression and immune system modulation |
| Morgell et al. [33], Sweden, 2021 | 16S rRNA PCR | Cystic fluid from 5 SCN 7 29 LGD 8-IPMN 8 HGD 9-IPMN 15 IPMN with associated PDAC |
PCR contamination prevention | Firmibutes, Proteobacteria, and Actinobacteria most common bacteria within pancreatic cystic fluid | Corroboration of previous results on pancreatic cystic fluid. Metabolomic characterization |
Despite substantial inter-individual variability of the gut flora, some studies concur in their findings, pointing at different bacterial species potentially involved in PDAC tumorigenesis.
The most prominent microbes identified in pancreatic tissue samples and associated with PDAC are Gram-negative bacteria, more specifically from the phylumProteobacteria[32][41][35,44]. directly compared pancreatic and fecal samples, showing especially forProteobacteriaan increased presence in the pancreas tissue. also detected increasedProteobacteriain human PDAC tissue [39][42] while Chakladar et al. showed an association for some members of the classesBetaproteobacteriaandGammaproteobacteriawith poor patient prognosis [32][35]. Of note, elevated levels ofProteobacteriawere detected in fecal samples of patients with PDAC [38][42][41,45], making stool analysis an attractive, cheap, and non-invasive method to detect intrapancreatic microbial shifts.
Some studies showed elevated levels of intratumoralEnterobacteriaceae, which also correlated with poor prognosis [32][34][35,37]. Moreover, another intestinal bacterium,Enterococcus faecalis, was identified within juice and tissue samples of PDAC-patients [37][40]. Since these bacteria are typical of the human gut, their presence within the pancreas could suggest a translocation from the gut.
Fusobacteriumspp. , a bacterial genus commonly present in the oral cavity during periodontal disease, was also found in PDAC tissue samples. However, so far, no data show an effect of its eradication, and neither genetic nor molecular PDAC patterns showed any correlation withFusobacteriumcolonization [30][35][33,38]. Similarly,P. gingivalishas also been detected in significantly higher concentrations within the pancreatic duct of periampullary malignancies [41][44] and in fluid of pancreatic cysts obtained through endoscopy [36][39].
Specific microbial signatures have also been associated with cystic precursor lesions and different PDAC tumor stages. observed that high-grade IPMN showed high levels ofFusobacterium nucleatumandGranulicatella adiacenscompared to non-IPMN cystic lesions [35][38]. Of note,Bacteroideswas also found in higher concentration in PDAC tumor samples [35][38], in particular the genusElizabethkingia[38][41]. These data suggest microbiome changes during tumor development.
Some microbiome patterns have also been described to act protectively. In particular, higher α-diversity (an indicator for bacterial variability) and higher levels ofSaccharoplyspora,Streptomyces, andPseudoxanthomonaswere associated with PDAC long-term survival [40][43]. In contrast, anaerobes likeLactobacillus, Roseburia, andFaecalibacterium, known to exert systemic anti-inflammatory effects, were significantly reduced in PDAC tissue [41][44].
Curiously, in one study the direct comparison of microbiota analyzed in tissue samples of healthy pancreas, chronic pancreatitis and PDAC did not show any differences between the samples [39][42].
Despite the large amount of published work, the question about the route by which these microorganisms reach the target organ is still not answered. Even though the oral administration ofSaccharomyces cerevisiaein mice was followed by a consistent presence of this microorganism within the main pancreatic duct [31][34], there is so far no proof of ductal migration of specific PDAC associated bacteria in humans.
3. The Role of the Microbiome in PDAC
The isolation of bacterial DNA directly from healthy and tumorous pancreatic tissue generates new research perspectives. On the one hand, it encourages identification of specific microbial signatures in the gut as novel non-invasive tumor markers, on the other hand, it adds a fascinating new player in carcinogenesis.
Most pancreatic cancers are believed to develop from non-invasive premalignant lesions, histologically defined as pancreatic intraepithelial neoplasia (PanIN). In these lesions, somatic mutations in genes like Kirsten rat sarcoma (KRAS) (codons 12, 13, 61) or, less frequently, guanine nucleotide binding protein (GNAS), are an early, almost universally found event [43][46]. Even though direct microbiota-associated tumor induction has not been described so far, some microorganisms have been observed to be associated to genetic alterations in PDAC (Figure 1).
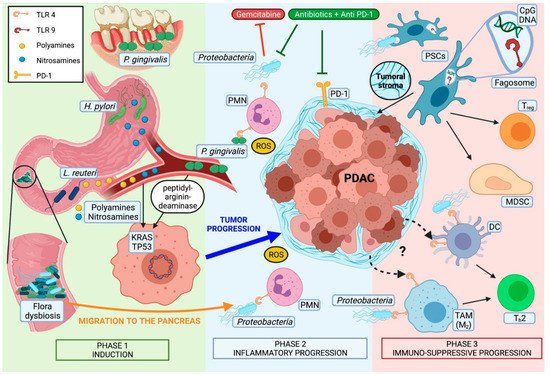
P. gingivalis, for example, can secrete peptidyl-arginine deaminase, an enzyme that is known to produce point mutations in tumor protein p53 (TP53) and KRAS [47][50].Toxypothrixsp. ,Acidovirax ebreusandShigella sonneialso correlate with the downregulation of signatures directly related to TP53 [32][35]. Obesity-induced alterations of gut microbiota could also play a role due to the higher incidence ofFirmicutesand the reduced numbers ofBacteroides[48][51] which lead to a pronounced production of deoxycholic acid, a bacterial metabolite known to cause point mutations [49][52].
The presence ofH. pyloriin human pancreatic cells has been associated with higher levels of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), activator protein (AP) 1, interleukin (IL) In this regard, a direct carcinogenic action ofH. pylorihas also been demonstrated in gastric cancer by deregulating polyamine metabolism and promoting oxidative stress [50][54]. Interestingly, increased serum concentrations of polyamines have also been found in mice and human PDAC subjects.
Endogenous carcinogens like nitrosamines have also been found in higher concentrations in in vivo models of PDAC. Their origin remains unclear. However, their extrapancreatic source and secondary transport to the target organ via bloodstream suggest a distant located microbial dysbiosis [51][56].
Of great interest is the recent discovery of epigenetic alterations in PDAC related toProteobacteria(likeAggregatibacter aphrophilusandAgrobacterium radiobacter), Gram-positive bacteria (likeBeutenbergia cavernae) andMycoplasma hypopneumoniae. These bacteria were strongly associated with an upregulation of specific methylation-related gene expression signatures [32][35].
Several preclinical models confirm the distant (gut) and local (intrapancreatic) role of microbiota in tumor progression.
Different mechanisms of microbiota-related tumor progression have been proposed over the last decade. The currently two most intensively debated hypotheses are cancer-associated inflammation and pro-tumorigenic immunomodulation within the tumor microenvironment (TME).
Oxidative stress disbalance is a pivotal mediator of inflammatory-induced carcinogenesis, and chronic inflammation has been recognized as a central facilitator in pancreatic carcinogenesis [52][57].
One of the proposed models sees a high-fat diet (HFD) as leading cause of an inflammatory response, which finally results in tumoral development of PanIN [53][58]. In a mouse model of colorectal carcinoma (CRC) More specifically, PDAC-cells exposed toE. faecalisshowed elevated expression of the pro-inflammatory cytokines CXCL8 and VEGF, that are known to promote fibrosis and angiogenesis. Additionally, colonization of PDAC byCitrobacter freundiiandPseudomonadales bacteriumhas been correlated with the upregulation of proinflammatory immune pathways such as the inflammasome [32][35].
Smoking has been characterized as a main risk factor for PDAC and it is further linked to bacterial dysbiosis [54][5]. Curiously, elevated levels ofA. baumanniiandM. hyopneumoniaefound on PDAC samples correlated with smoking-mediated changes in the genome that cause pancreatic cancer [32][35].
Nutritional habits influence the microbial composition. Beneficial species of the gut flora likeRoseburiaandEubacterium rectalewere decreased by low-carbohydrate and high-protein diets [55][62]. Same dietary regimens are related to reduced intestinal butyrate levels, a short-chain fatty acid involved in cell differentiation, apoptosis, and histone hyperacetylation, all effects thought to be associated with carcinogenic processes [55][62]. In contrast, high energy diets can cause the activation of pattern recognition receptors (PRRs) like Toll-like receptor type 4 (TLR4) by facilitating the absorption of bacterial lipopolysaccharide (LPS) in the gut.
These observations suggest a role for intestinal dysbiosis in facilitating an aspecific inflammatory status that has its origin in external factors, like dietary habits, and exerts its effect in the gut and in more distant locations like the pancreas.
Microbial dysbiosis, acting remotely (gut) and locally (intrapancreatic), has been associated with a TME shift towards an immunotolerant phenotype. In particular, Chakladar et al. demonstrated thatC. freundiiandM. hyopneumoniaecorrelate with multiple immunosuppressive pathways [32][35]. Some studies even consider the microbiota as a new component of the TME [56][64].
A role of the microbiota acting remotely from the gut has been recently demonstrated in heterotopic mouse xenografts. Bacterial depletion resulted in an increased expression of tumor suppressor genes death-associated protein kinase 2 (DAPK2), Krüppel-like factor 9 (KLF9), and Lumican (LUM) while microbiota-intact mice showed upregulation of pro-tumorigenic genes tenascin C (TNC), chemokine (C-X-C motif) ligand 10 The immune status of the TME within the PDAC differed significantly, depending on the presence or absence of the intestinal microbiota. Mice lacking adaptive immune system (non-obese diabetic–severe combined immunodeficiency: NOD-SCID) had increased CD45+innate immune cells in their PDAC xenografts only if treated with wide-spectrum antibiotics.
In a heterotopic mouse model of PDAC, the antibiotic-driven depletion of gut bacteria resulted in increased numbers of anti-tumorigenic lymphocytes, like CD3+CD4+IFNγ+, CD3+CD8+IFNγ+, CD3+IFNγ+, and reduced occurrence of pro-tumorigenic CD3+IL-17+and CD3+CD4+IL-10+cells [57][65]. Similarly, mouse models of slow progressive PDAC (p48cre; LSL-KrasG12Dknown as KC-mice) and PDAC xenografts also showed suppression of the intratumoral adaptive-immunity cells [38][58][41,66]. In contrast, germ-free KC-mice showed higher intratumoral anti-tumorigenic lymphocytes. However, after stool transplantation from mice harboring an aggressive form of PDAC (Pdx1cre; LSL-KrasG12D;Tp53R172H, known as KPC-mice), anti-tumorigenic lymphocytes were significantly reduced [59][67].
However, available data suggest an essential role of the TLRs in pancreatic tumorigenesis. Its increased presence on murine and human PDAC tumor cells and macrophages [38][44][60][41,47,68] also suggests a crucial role as intratumoral immunomodulators. In that context, inhibition of the TLR-associated myeloid differentiation primary response 88 (MyD88) pathway by intrapancreatic dendritic cells (DC) resulted in a pro-tumorigenic, fibroinflammatory environment, with consequent Th2-shift and acceleration of the transition from pancreatitis to carcinoma [44][47].
Activation of TLR2, TLR4, and TLR5 was observed to be higher expressed in mice exposed to a cell-free extract of gut bacteria-derived from KC-mice compared to wild-type (WT) mice [38][41]. Other studies showed microbiota-induced activation of TLR4 and TLR7 resulting in pro-tumorigenic immunosuppressive TME in early and progredient tumor stages [44][46][61][47,49,70] TLR5 has been described to be upregulated in TME macrophages, and to be related to tumor growth [38][41]. Finally, TLR9 activation has been demonstrated to induce pancreatic stellate cells (PSCs) of the tumoral stroma to become fibrogenic and to attract Tregcells and myeloid-derived suppressor cells (MDSCs) [46][49]
Regarding TLR2, the findings are less clear. While high expression and activation on tumor-associated macrophages (TAM) is related to tumor growth in a PDAC mouse model [38][41], TLR2-agonists have been shown to be an effective adjuvant immune-therapy against PDAC [62][71]. Moreover, tumor progression due to intracellular presence ofP. gingivalisunder hypoxic conditions has been demonstrated to be independent from TLR2 signaling. On the contrary, in the context of oral carcinomaP. gingivalispromotes tumor growth in a TLR2-dependent manner [14][17].
Some TLRs have been already addressed as potential targets for immunotherapy in pancreatic cancer. As already reported, synthetic high affinity TLR2 agonists have been observed to induce boost immunity when given as vaccine adjuvants in murine PDAC models [62][71]. In a phase I/II, trial patients with incompletely resectable PDAC received during surgery an intratumoral injection of MALP-2, a synthetic lipopeptide which activates the immune response through TLR2/6. Antitumoral effects like inhibition of stromal proliferation have been observed in murine PDAC models following stimulation as well as following inhibition of this receptor [60][63][68,73].
Taken together, these findings show a complex interaction between microbiota and intrapancreatic immune cells in the context of PDAC. Even though we are far from fully understanding which specific pathway and molecular signaling are involved in establishing this intratumoral immunotolerant phenotype, microbiota have been identified as an important player in this setting. Some authors propose a Janus-faced involvement of TLRs. While the peripancreatic pro-inflammatory response might be only the first effect of TLR-activation, thanks to molecular feedback control mechanisms the same receptors could for a second time modify their signaling and switch from a Th1 to Th2 response with immunosuppressive features
The complex interactions between microbiome and immune system also seem to influence prognosis and treatment response to different adjuvant therapies.
Concerning the influence of systemic treatments in a human study, the ablation ofKlebsiella pneumoniae, which may promote gemcitabine resistance in PDAC, has been associated with improved survival [64][76]. Similarly, bacterial ablation enabled checkpoint inhibitors’ efficacy by upregulating programmed cell death protein 1 (PD-1) expression in PDAC mouse models [38][41] Of note, in renal and non-small cell lung carcinoma, bacterial ablation reduced the effectiveness of checkpoint blockade therapy [65][77]. Hence, a selective antibiotic ablation combined with other systemic treatments could represent a favorable strategy, especially if one bears in mind that human PDAC tissue has been demonstrated to harbor predominantly Gram-negative bacteria [38][64][41,76].
While genomic analyses focus on comprehensive bacterial species profiling, new functional tools like proteomics and metabolomics address dysbiosis from another perspective. These studies concentrate their efforts on the harmful protein microenvironment caused by the dysbiosis.
One of the first conducted metabolomic studies focused on the presence of elevated serum polyamine levels in a genetically engineered PDAC murine model (KPC-mice) as well as in human PDAC samples. Polyamines are of known bacterial origin. Interestingly, they emerged in KPC-mice already early before any detectable tumor was located and were also present in human serum samples of PDAC patients. These data suggest early changes in the gut flora in patients developing PDAC and could therefore represent a new, early non-invasive marker [66][55].
Recently, metabolomics analysis has been also performed on serum and cystic fluids of patients with neoplastic and non-neoplastic cystic lesions of the pancreas. Among them, PDAC precursors, showed significant correlations with defined metabolic patterns, like purine oxidation, heme metabolism, acyl-carnitines and glycolytic metabolites. Furthermore, absolute quantitative measurements on cyst fluid highlighted acyl-carnitines as the top discriminants between neoplastic and non-neoplastic cystic lesions. The observed correlation between 16S RNA copy numbers and metabolite levels stresses the microbial origin of this metabolic “signature” [33][36].
Of note, proteomic bacterial profiling of bile from patients bearing PDAC showed overexpression of IL-8, which is known to be stimulated by bacterial biofilm formation. The authors of this study also observed elevated levels of primary and secondary compounds, which play an important role in biofilm formation and act as inhibitors for concurrent bacterial species, suggesting the presence of major competition among different bacteria in this context [67][78].
4. The Role of the Mycobiome in PDAC
The role of microbial components other than bacteria in tumorigenesis is quite unexplored [68][79]. The fungal component is known as “mycobiota”, with the term “mycobiome” indicating their collective genomes. Fungi are estimated to comprise less than 1% of all commensal species [69][80].
Recently, mycobiome alterations have also been observed in the context of human malignancies like CRC and PDAC, with the intrapancreatic mycobiome of PDAC patients clustering differently from that of healthy individuals [31][34]. PDAC and CRC showed elevatedBasidiomycotalevels likeMalasseziaspp. , whileAscomycotawere reduced. In particular,Malassezia globosashowed good accuracy in differentiating CRC and PDAC from healthy controls [31][70][34,81].
Interestingly,Malasseziawas also the most prevalent genus in the pancreas of KC-mice. In this murine PDAC model, levels increased in parallel with tumor growth, reaching its peak when tumor development was completed. Interestingly, in earlier life stages wild-type and KC mice do not differ in their mycobiome, pointing at fungal dysbiosis and especially atMalasseziaas a crucial player in PDAC development [31][34].
Malasseziais commonly found on the skin with the capability of gut colonization [71][82]. It encodes some secreted enzymes similar toCandida albicans, which have also been described to contribute to carcinogenesis [72][73][83,84].
Its contribution to PDAC progression seems to be related to the presence of mannose-binding lectin (MBL). Higher MBL expression was associated with worse survival in PDAC patients. In contrast, in MBL-null miceMalasseziadid not accelerate tumor progression, nor did the treatment with amphotericin B protect from tumor growth in these mice [31][34].
MBL is a soluble PRR, which recognizes among other pathogenic carbohydrate antigens fungal pathogens and activates the lectin pathway of the complement cascade [74][85]. This results in the production of C3a. The oncogenic activity of C3a has been demonstrated in mice with tumor growth mitigation due to deficiency of C3 or its receptor, suggesting a crucial role of this pathway in tumor development [31][34].
A complement-driven tumor progression has also been observed in human specimens [75][86]. C3a has been classified as potentially oncogenic since it could increase proliferation, motility, and invasiveness of tumor cells. Signaling of complement receptor C3a (C3aR) has been shown to be involved in epithelial–mesenchymal transition (EMT), while C5a acts in an immunosuppressive way by inducing apoptosis of CD8+cytotoxic cells, by attracting MDSC into the tumor, and by participating in the shift of the macrophages towards an M2-phenotype [76][87]. Furthermore, the expression of C3a was also associated with reduced survival of PDAC patients [31][34].
Of note, high levels of CD59, an inhibitor of the membrane attack complex (MAC) of the complement, have been observed on pancreatic cancer cells. This could explain why in the context of PDAC only the tumorigenic effects of the complement are present, while the lytic activity of the MAC seems to be suppressed. Moreover, the expression of CD59 appears to be induced by alternatively activated macrophages [77][88], whose polarization could be influenced by DCs and Dectin-1 activation (Figure 2).
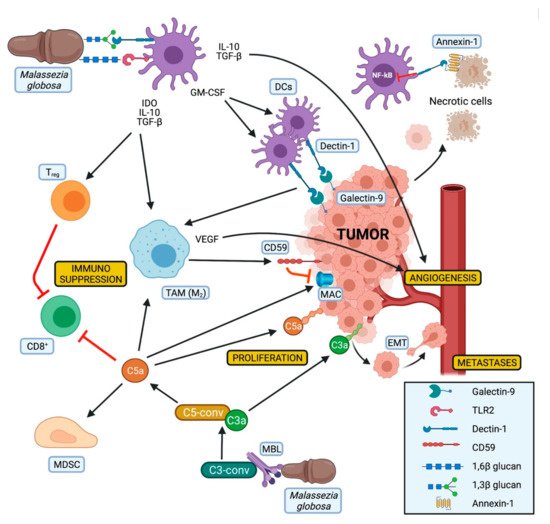
To the best of our knowledge, no treatment targeting the MBL pathway has been described so far in a preclinical and/or clinical PDAC setting.
Dectin-1 is another fungal PRR that has been described as an emerging player in pancreatic oncogenesis. Dectin-1 is a non-classical C-type lectin receptor generally expressed on the surface of myeloid-monocytic cells and some T cells that recognize β-1,3 and β-1,6 glucan polysaccharides expressed mostly by yeasts and fungi, with its activation resulting in NF-kB expression [78][89]. This receptor has been recently observed on PDAC and tumor-infiltrating myeloid cells of human and murine tissues [80][91].
Dectin-1 can be activated by fungal glucans leading to the activation of the innate immune system and to the expansion of DCs through GM-CSF production [78][89]. In addition, Dectin-1 expression on DCs and macrophages is critical for natural killer cells mediated elimination of tumor cells expressing N-glycan structures [83][94]. On the other hand, the interaction of Dectin-1 with Galectin-9, a lectin with an affinity for β-galactoses highly expressed on PDAC cells, leads to CD8+T cell exhaustion via T-cell immunoglobulin domain and mucin domain Another study observed Dectin-1 as tolerogenic receptor for annexins, proteins expressed on apoptotic cells, which can induce immune tolerance via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase-2 and can also inhibit the NF-kB pathway [81][82][92,93] (Figure 2).
As already mentioned, TLR2 has been related to PDAC. Fungal presence within PDAC tissue together with elevated expression of Dectin-1 and TLR2 suggests crosstalk between mycobiota and cancer cells. However, the function of TLR2 is not clearly defined. On the one hand, TLR2 activation has been related to tumor growth [38][41]; on the other hand, TLR2-agonists act antitumorigenic inducing apoptosis on PDAC cells [62][71].
Despite the Janus-like observations about the isolated Dectin-1 and TLR2 activation, synergisms between these two receptors have been described when binding β-glucan [84][96] (Figure 2). SinceMalasseziacontains both β-1,3 and β-1,6 glucan, it could be suggested that those two molecules could, respectively, bind Dectin-1 and TLR2, finally leading to their synergistic activation. In a mouse model of type-1 diabetes this synergistic activation resulted in the expression of immunoregulatory cytokines like IL-10 and TGF-β as well as indoleamine 2,3-dioxygenase (IDO).
TLR9 also needs to be considered in the context of mycobiome related PDAC development, since it has been linked to induction of stroma-producing PSCs, Tregand MDSCs [46][49] and its expression on macrophages can be stimulated by different fungi, includingMalassezia[85][97].
In different mouse models, the ablation ofMalassezia globosawith amphotericin-B was protective against tumor progression. Of note, the recolonization of amphotericin-B pretreated The same did not happen by recolonization with other fungi, likeCandidaspp. Besides, amphotericin B gavage in germ-free, tumor-bearing mice did not influence tumor progression at all [31][34].