Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Bruce Ren and Version 1 by Alfonso Urbanucci.
The dysregulation of chromatin and epigenetics has been defined as the overarching cancer hallmark. By disrupting transcriptional regulation in normal cells and mediating tumor progression by promoting cancer cell plasticity, this process has the ability to mediate all defined hallmarks of cancer.
- prostate cancer
- epigenetics
- chromatin
- lineage commitment
- lineage plasticity
- chromatin-associated factors
- castration resistant prostate cancer
- drug resistance
- androgen receptor signaling inhibitors
- chromatin regulators alterations
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Chromatin structure and epigenetics are intertwined but, nonetheless, distinct entities that have been implicated in prostate cancer (PC) disease initiation and progression. Here, we consider epigenetics to be the collection of DNA modifications such as DNA methylation. This definition of epigenetics can also include histone modifications and the binding of transcription factors (TFs) to DNA. Histone modifications are often referred to as epigenetic profiles as they determine chromatin states and nucleosome positioning, which, in turn, allows for DNA accessibility (Figure 1). Therefore, we additionally define chromatin structure as histone modifications and nucleosome positioning, as well as the three-dimensional (3D) organization of the chromatin within the nucleus (Figure 1).
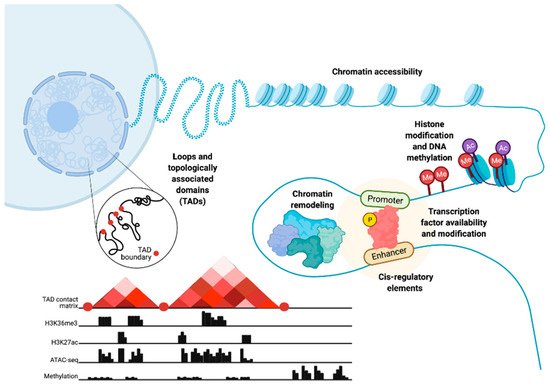
Figure 1. Epigenetic- and chromatin-related mechanisms with potential for dysregulation in prostate cancer cells. Epigenetic dysregulation can occur at multiple levels, including changes in chromatin accessibility, histone and DNA modification through processes such as methylation, chromatin remodeling, modification of transcription factors and changes in their availability, cis-regulatory elements, chromatin loops, and topologically associated domains. These chromatin and epigenetic features can be analyzed via the integration of multiple high-throughput sequencing data types. These data include chromatin conformation capture (Hi-C) to understand the 3D chromatin structure and topologically associated domains, chromatin immunoprecipitation (ChIP-seq) to study histone markers, assay for transposase-accessible chromatin (ATAC-seq) to show chromatin accessibility patterns, and DNA methylation sequencing. Figure created with BioRender.com.
PC continues to be a major cause of cancer-related death in men worldwide [1]. Although primary intervention with radiotherapy or surgery and androgen deprivation therapy (ADT) have a curative intent in hormone-sensitive PC (HSPC), metastatic disease remains incurable, despite the introduction of combination approaches [2]. Targeted systemic therapies with androgen receptor (AR) signaling inhibitors (ARSIs) such as abiraterone or enzalutamide are primarily used to treat relapsed castration-resistant prostate cancer (CRPC) [3]. Employment of these agents has been shown to be effective, especially in high risk primary metastatic HSPCs patients, and their use is becoming more common [4,5][4][5]. Although these combination approaches have demonstrated survival benefits, they have also been shown to contribute to the emergence of more aggressive castration-resistant tumors [6]. The majority of castration- and some ARSI-resistant PCs are characterized by increased AR signaling [7,8][7][8]. However, alternative forms of castration-resistance have also been identified, including forms that are AR-negative with neuroendocrine-like features [9], forms indifferent to AR [10], and forms that are more dependent on alternative signaling pathways such as fibroblast growth factor (FGF) [11]. Additionally, forms of castration-resistance dependent on other TFs such as the glucocorticoid receptor (GR) or the pluripotent stem cell TF SOX2 [12,13,14,15][12][13][14][15] have been described.
Along with others, we previously reported that the emergence of castration-resistance and AR overexpression are associated with chromatin reprogramming [16,17,18,19][16][17][18][19]. As with other malignancies, PC is thought to arise from and be driven by oncogenic genetic alterations. However, as with many cancers, PC cannot be explained solely on the basis of genetic alterations [20]. PC in particular has a relatively low mutational load at presentation [21] and prostate carcinogenesis is not clearly driven by any particular genetic alterations [22]. Nevertheless, HSPCs and CRPCs are characterized by the typical cancer hallmarks [23,24][23][24] that mediate carcinogenesis, disease progression under treatment pressure, and cancer growth beyond the tumor microenvironment (TME).
Flavahan and colleagues first proposed the concept of epigenetic plasticity, by which alterations in the structure of the chromatin, or chromatin states, and epigenetic alterations would be able to confer the full range of cancer hallmarks by altering transcriptional regulation [25,26][25][26]. Indeed, many of the epigenetic and chromatin regulators that drive normal tissue development and differentiation are well-known oncogenes and tumor suppressors recurrently mutated or aberrantly expressed in different malignancies.
Similarly, it is well known that cellular lineage identity is defined by tightly regulated chromatin-related processes and the epigenetic landscape [27]. The concept of lineage plasticity is of clinical relevance for PC as it is a common mechanism of resistance following the increasing usage of more potent ARSI in primary disease [11,28][11][28].
Due to the dynamic sets of chromatin and epigenetic alterations and their reversibility, such alterations can play a significant role in driving both carcinogenesis and progression to treatment-resistant disease. These alterations can also be stochastically different in individual cells [25]. Therefore, epigenetic plasticity can be the basis of the heterogeneity observed among PC patients, but also between different tumor foci of the same prostate [29,30,31][29][30][31]. It is apt that epigenetic plasticity can consist of genetic changes that allow normal cells to transform through alteration of lineage commitment and further allow malignant cells to adapt under diverse treatment pressure by mediating lineage plasticity.
In this review, we highlight the contribution of chromatin- and epigenetics-related processes to prostate carcinogenesis and progression to treatment resistance. We explore the role of epigenetic regulation and TFs in lineage commitment and plasticity in the normal prostate and PC cells, the mutations and the altered expression of key genes coding for chromatin-associated proteins, alterations in DNA methylation patterns, and changes in the structure and 3D organization of the chromatin (Figure 2).
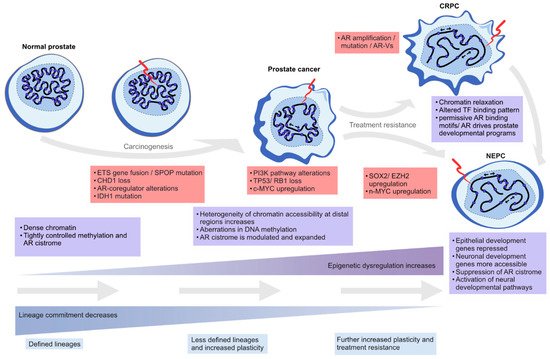
Figure 2. Epigenetic plasticity in prostate cancer. Epigenetic dysregulation (light blue boxes) is in the forefront of lineage plasticity as well as in carcinogenesis and therapy resistance. Normal prostate epithelium is renewing at a steady state as terminally differentiated luminal cells are slowly replaced by progenitor cells. As genetic alterations accumulate due to cell division and the normal aging process, driver alterations (red boxes) such as ETS gene fusions or SPOP mutations emerge. The mutational processes lead to less ordered chromatin structure, as characterized by chromatin relaxation at distal regulatory regions, alterations in DNA methylation and histone modifications, and dysregulation of higher order chromatin structures, which alters the binding of key TFs such as AR. Some cells gain stem-like properties, leading to increased proliferation capacity and reduced apoptotic rates, which leads to tumor formation over time. The plasticity of the cellular identity is also in a key role during the emergence of treatment resistance as the cancer cells can repurpose differentiation-promoting transcription factors such as AR into regulatory regions supporting developmental gene expression (seen in castration-resistant prostate adenocarcinoma, CRPC), or transdifferentiate into non-luminal, small cell prostate carcinoma or neuroendocrine prostate cancer (NEPC).
2. Prostate Lineage Commitment and Prostate Cancer Plasticity
Here, we collect evidence of the involvement of lineage commitment and cancer cell plasticity in PC initiation and resistance to treatment, respectively.
The normal epithelium of the prostate secretory acini is composed of a layer of luminal cells oriented towards the acinar lumen and a basal cell layer that also includes rare neuroendocrine cells. The determinants of prostatic epithelial differentiation include master regulators such as AR, FOXA1, and NKX3-1. Talos and colleagues showed that these TFs were sufficient and required for differentiation of iPS cells of mouse fibroblast origin in prostatic cells engrafted in the renal capsule [32]. Xie et al. showed that AR may also be required in the basal-luminal intermediate cells to produce fully differentiated luminal progeny in adult mice [33]. The same study found that AR expression is not needed for survival of the luminal cells but is essential for normal luminal differentiation and morphology [33]. This may be, in part, due to an indirect effect by NKX3-1, as it is an AR target gene and was not expressed after deletion of AR [33]. Dutta et al. had previously shown that NKX3-1 is a prostate-specific master transcriptional regulator that can transdifferentiate the seminal vesicle epithelium into prostate epithelium [34]. Of note, the seminal vesicle epithelium expresses AR and FOXA1 following the introduction of NKX3-1 [34]. Differentiation by NKX3-1 was found to be mediated by histone demethylase UTY and histone methylase G9a [34]. The stromal component seems to contribute to the re-differentiation process as the urogenital sinus mesenchyme is always co-transplanted with the epithelial cells in these experiments [32,34][32][34].
AR and FOXA1 have been found to also be important for the regulation of the Homeobox (HOX) A genes [35]. The HOX genes regulate prostate development in mice [36] and the paralogous HOX13 genes (HOXA13, HOXB13, and HOXD13) are still expressed in the luminal epithelium of the human adult prostate [36]. In recent single cell analyses, Guo et al. showed that terminally differentiated luminal cells express NKX3-1 and HOXB13 together with AR [37].
Experiments in mice have shown that the prostate epithelium displays a regenerative capacity following repeated cycles of androgen deprivation [38,39][38][39]. This led to the hypothesis that the prostate epithelium may also harbor stem cells responsible for tissue renewal [38]. Recent advances in single cell sequencing have facilitated a more detailed characterization of the complexity of prostate cell types without having to rely on restricted sets of cell surface markers for their classification [40]. These single cell approaches have led to the identification of club cells (KRT5−, KRT8−, and SCGB1A1+) and hillock cells (KRT5+, KRT14−, KRT13+) with stem cell potential [40]. Similar to lung club and hillock cells, progenitor-like cells that are able to differentiate into goblet cells and ciliated cells, these newly described prostate cells are efficient at reconstituting the prostate epithelium in in vivo studies following androgen deprivation, as shown by Karthaus et al. [41], although their transdifferentiation to basal cells is rather limited [40,41,42][40][41][42].
Maitland and Collins have reviewed how the prostate epithelium constantly renews and the terminal differentiation of luminal cells is associated with a higher rate of apoptosis [43]. The existence of intermediate cells, which are cells with both basal and luminal features in the prostate epithelium, has been acknowledged for a long time [44]. In lineage tracing studies with mice, both basal and luminal cells have been shown to contribute to the renewal of prostate epithelium during androgen deprivation and add back cycles, indicating that there are several degrees of stemness within the adult prostate epithelium [37,41,45][37][41][45]. In previous studies, both luminal and basal stem or progenitor cells have been shown to act as the PC-initiating cells, but PC is generally characterized by the absence of basal cells [45,46,47,48,49,50][45][46][47][48][49][50].
Lineage plasticity refers to the reversal of the process of lineage commitment, either by dedifferentiation of the more differentiated cells or, in extreme cases, transdifferentiation directly (or via an intermediate) to another epithelial cell lineage. This is a process that is unlikely to occur in the normal prostate under physiological conditions. However, pathological processes or stressors such as inflammation may alter this scheme [51]. Alterations in key epigenetic- and chromatin-associated or environmental stress factors can disrupt the epigenetic homeostasis of normal cells, which may lead to lineage plasticity, differentiation arrest, and accumulation of undifferentiated cells in transition. The role of the TME in determining such alterations is not well known, but factors such as hypoxia [52] and other metabolic stressors [53] have been associated with more aggressive PC phenotypes. A well-established example of metabolic stress leading to epigenetic changes is the dysregulation of one-carbon metabolism and its effects on both DNA and histone methylation [53,54][53][54].
Lineage plasticity during prostate carcinogenesis is poorly understood. However, for example, the upregulation of c-MYC is a common early event [55] and has been implicated in the gain of stem cell properties and repression of differentiation [56]. Normal luminal cells repress MYC expression via AR/β-catenin/TCF-4 signaling in the presence of androgens, leading to growth arrest, but overexpressing c-MYC rescues cell proliferation [57,58][57][58]. This highlights the opposing roles of AR as the main differentiation factor and growth suppressor in the non-transformed secretory luminal prostate cells and as a prominent driver of PC cells’ proliferation, as discussed below.
Cells with regenerative potential within the normal prostate tissue have been hypothesized to be the cells of origin for PC [59]. Indeed, Song et al. recently described club-like PC cells in primary PC specimens [60]. These cells are transcriptionally similar but have higher AR expression and an enhanced androgen signaling signature when compared to club cells from normal prostates [60] (Table S1). The overexpression of AR in these cells is consistent with the modulation and expansion of the AR cistrome, which is a well-documented feature of PC initiation [61]. Under this scenario, AR overexpression induces changes in the AR transcriptional program, leading to cell survival and proliferation, possibly via the alteration of the pioneer activity of cooperative TFs of the AR such as HOXB13 [61]. Interestingly, recent analyses with mice expressing the F133V mutant form of Speckle Type BTB/POZ Protein (SPOP) showed that this mutation is sufficient to modify chromatin accessibility and binding of AR and FOXA1 at PC specific genomic sites [61,62][61][62]. Further expansion of the AR cistrome has also been reported during progression to CRPC [63,64][63][64].
The role of some luminal progenitor cells as possible cells of origin for PC is also supported by studies in Tmprss2-CreER;Ptenflox/flox mice [37]. The TACSTD2/Trop2 (encoded by Tacstd2)-expressing luminal progenitor cells characterized by Guo et al. have similar transcriptional features to the club cells characterized by Henry et al., Karthaus et al., and Song et al., based on the presence of markers such as PIGR, PSCA, and KRT4 [37,40,41,60][37][40][41][60] and high expression of TACSTD2 [41,60][41][60] (Table S1). Similarly, Kwon et al. found that TACSTD2+ luminal prostate cells were more efficiently transformed in vitro in an organoid-forming assay than TACSTD2−luminal cells [65]. The authors also showed that TACSTD2+ luminal cells express SOX2 and display remarkable plasticity by transdifferentiation to de novo neuroendocrine PC (NEPC), even in the absence of selection pressure from treatment with antiandrogens [65].
Although the above-described studies suggest that multiple subpopulations of cells can give rise to PC [37,45[37][45][46][47][48][50],46,47,48,50], their transcriptional features seem to converge toward features of luminal progenitor or club-like cells [37,41,48,49,60][37][41][48][49][60]. Undoubtedly, the cell type of origin of PC can have clinically relevant consequences in terms of tumor trajectory and prognosis [50].
Treatment of metastatic HSPCs with ARSI in combination with ADT imposes a strong negative selection pressure on the cancer cell population. Multiple treatment resistance mechanisms arise from genetic and epigenetic alterations, leading to a rewiring of alternative bypass pathways driving tumor cell growth [66], but the majority of CRPCs remain AR-dependent through maintenance of AR signaling [67]. This imposes chromatin reconfiguration and epigenetic plasticity in ARSI-resistant PC cells, leading to heterogeneous CRPC phenotypes more or less dependent on AR signaling, with both luminal and basal features [68,69,70][68][69][70]. A recent single cell analysis by He and colleagues showed that different AR transcript variants are ubiquitously expressed prior to treatment with ARSI [71]. They also found that ARSI drives resistance pathways such as epithelial to mesenchymal transition (EMT), a form of lineage plasticity generally associated with metastatic disease [71].
Chromatin reprogramming is a key feature of lineage plasticity during treatment resistance, for example by allowing reactivation of pathways that are normally active only during prostate tissue development [19,64,72][19][64][72]. Zhang et al. found that loss of the chromatin-modifying helicase CHD1 confers enzalutamide resistance by inducing marked changes in chromatin accessibility, along with transcriptional rewiring and upregulation of TFs NR3C1 (GR), POU3F2 (BRN2), TBX2, and NR2F1, leading to gene expression changes including a reduction in luminal and an increase in EMT markers [68].
Cellular plasticity has also been associated with alterations in tumor suppressor genes and chromatin reconfiguration, exemplified by the deletion of TP53 and RB1 that can lead to treatment resistance by allowing for diverse transcriptional programs [6,69,70][6][69][70]. P53 and pRb (encoded by TP53 and RB1, respectively) cooperate to suppress expression of SOX2, a well-known factor of pluripotency, which, in part, explains the association of these factors with lineage plasticity [15,73][15][73]. Using an isogenic model of HSPC (LNCaP) and CRPC (C4-2), Mandingo et al. showed that RB1 loss leads to a reconfiguration of E2F activity for increasing the production of antioxidants, protecting cells against doxorubicin in the CRPC cells that is not observed in the HSPC cells [74]. These findings highlight how transcriptional reprogramming by the same TF can be modulated in different disease stages [74].
In the context of ARSI resistance, so-called treatment-induced neuroendocrine prostate cancers (t-NEPCs) (reviewed by Rubin et al. and Kaarijärvi et al. [28,75][28][75]) represent a tumor phenotype mediated by chromatin and transcriptional plasticity, which, in turn, drives cellular plasticity. During therapies with ARSIs, AR-dependent cells are depleted, while transcriptionally and epigenetically heterogeneous AR-indifferent cells increase their growth and survival potential [11,68,69][11][68][69]. Increased mutation frequency in tumor suppressor genes such as TP53 and RB1 suggests that t-NEPCs require these alterations for tumor selective reprogramming [6,15,73,76][6][15][73][76]. The t-NEPC tumors acquire neuroendocrine features and suppress luminal transcriptional programs via the upregulation of SOX2 and EZH2 in mouse models [15,73][15][73] and the upregulation of LHX2 and ISL1 in human cells, which goes hand in hand with increased chromatin accessibility and the transcriptional output of neuroendocrine lineage-specific genes [76]. FOXA1, a well-known AR pioneering factor and a mediator of the transcriptional output in prostate adenocarcinoma, has also been shown to mediate NE-specific transcription in NEPC [77]. NE-specific genes are repressed in normal differentiated prostate epithelial cells, but during transdifferentiation following the activation of NE TFs such as ASCL1 and NKX2-1 in these cells, the chromatin structure in their proximal regulatory regions is rewired and allows for their expression [77]. The loss of the transcriptional repressor REST, which normally represses ASCL1 and NKX2-1, has been implicated in this process [78]. Active AR signaling keeps REST stable via inhibition of E-ubiquitin ligase β-TrcP, so ARSIs may contribute to the downregulation of REST [79].
Ultimately, PC progression is a continuous process resulting from the selection pressures of treatment upon the tumor, the TME, and the host immune system [80].
3. Mutations and Expression Dysregulation of Genes Coding for Chromatin-Associated Factors
Many studies have illustrated how chromatin-associated factors affect both lineage commitment and lineage plasticity, which is likely to drive tumor progression [64,68,77][64][68][77]. PC cell lineage plasticity is driven by genetic alterations, gene expression changes, and the altered activity of chromatin-associated and epigenetic regulators [6,25,81,82,83,84][6][25][81][82][83][84]. These chromatin-associated factors can be broadly categorized by their protein function into TFs, transcriptional co-regulators, chromatin modifiers, and genes involved in mRNA transcript synthesis or processing. In this section, we performed an analysis of their mutational and expression patterns in prostate carcinogenesis and the development of treatment resistance. To gain a comprehensive view on chromatin-associated factors, we utilized a list of 2754 genes previously annotated to the above-mentioned functional groupings [85] and queried their alteration status in publicly available PC patient datasets.
Somatic mutation data from the International Cancer Genome Consortium (ICGC) prostate adenocarcinoma datasets [86] showed that only 34 were genes mutated in more than 1% of patients with potential protein function-altering effects, reflecting the overall low mutation frequency of these tumors (Table S2). As the ICGC cohort largely consists of early stage primary prostate tumors, we repeated the analysis using two metastatic CRPC patient cohorts from Robinson et al. and Grasso et al. to include 150 pre- or post-ARSI mCRPC biopsies [87] and 50 heavily pre-treated lethal CRPCs exposed only to first generation ADT [88], respectively. We identified an additional 78 genes coding for chromatin-associated proteins in more than 2% of these patients with protein-altering mutations (Table S2). Altogether, 18 genes, including TP53, FOXA1, SPOP, and CDK12, were recurrently mutated in both the ICGC early stage and advanced CRPC tumors. The proportions of mutated genes from different functional categories did not differ significantly in the early versus treatment-resistant disease stages.
We further assessed the expression of the 94 recurrently mutated genes (Table S2) during prostate carcinogenesis and the development of treatment resistance in a previously published RNA-sequencing dataset of benign prostatic hyperplasia (BPH, n = 10), untreated PC (n = 16), and CRPC (n = 11) samples (Figure 3) [89]. We observed expression changes in several known PC driver genes including the loss of expression of TP53 and SPOP during the transition from untreated PC to CRPC, as well as the increased expression of AR, BRCA2, KDM6A, and CDK12 in CRPC. Interestingly, TFs that were mutated in PC and CRPC or only in CRPC showed higher expression in those categories, with the exception of decreased TP53 expression in CRPC, which is potentially a consequence of its wide-ranging tumor suppressive functions [70]. TFs mutated only in CRPC typically had lower expression in CRPC, with the exception of AR and EHMT1, a histone methyltransferase with potential tumor suppressive function in the prostate [90]. Nearly all recurrently mutated chromatin modifiers had the highest expression in CRPC irrespective of the mutation frequency (Figure 3). Among the genes involved in epigenetic, chromatin, or gene regulation that were not recurrently mutated, 68 (2.6%) genes were significantly upregulated during prostate carcinogenesis (PC vs. BPH), while 80 (3.0%) genes were upregulated during the development of treatment resistance (CRPC vs. PC). In contrast, 34 (1.3%) non-recurrently mutated genes were downregulated during prostate carcinogenesis and 77 (2.9%) were downregulated during treatment resistance. TFs were the most common group of genes to be aberrantly expressed, particularly amongst the genes upregulated during carcinogenesis (46 of 68 genes, 68%) or downregulated during carcinogenesis (18 of 34 genes, 53%).
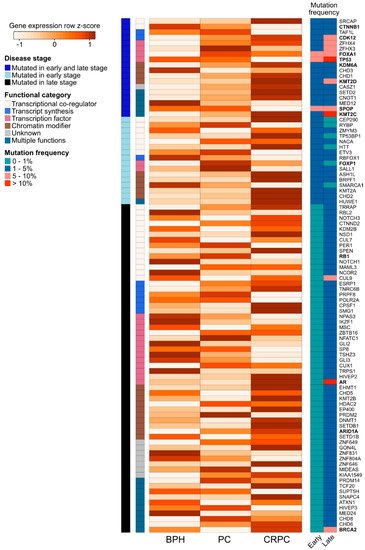
Figure 3. Expression changes of 94 recurrently mutated genes coding for chromatin-associated proteins in prostate carcinogenesis and development of treatment resistance. Row-scaled log2 mean expression values for each gene (the rows) are shown in a heatmap for benign prostatic hyperplasia (BPH), untreated prostate cancer (PC), and castration-resistant prostate cancer (CRPC) patient samples. The names of frequently studied genes are shown in bold. Each row is annotated with the disease stage, in which the gene is found to be recurrently mutated (either early stage (PC), late stage (CRPC), or both early and late stage). The rows are also annotated with the functional category of each gene. On the right, two columns show the mutation frequency of the gene in early- and late-stage disease in four categories (0–1%, 1–5%, 5–10%, and >10%).
Our analysis further highlighted alterations in groups of genes with functions related to PC lineage plasticity. These included the master transcriptional regulators FOXA1 and AR [91,92,93,94][91][92][93][94], the KMT2A-D transcriptional co-activators involved in development [95], and a number of chromodomain (CHD) genes, including CHD1, involved in chromatin remodeling and transcription activation [96]. Loss of CHD1 has been reported in 15% of HSPCs and 17% of CRPCs [97] and CHD1 loss has been implicated in PC cell chromatin rewiring with tumor-suppressing functions [68] and increased sensitivity of PC tumors to DNA damage [98]. The simultaneous dysregulation of tumor suppressors such as TP53 and RB1 in our recurrently mutated gene list has also been shown to promote PC plasticity [99].
In addition to genetic variation in coding regions as explored here, recent studies have also shown that alterations within noncoding regions affect chromatin conformation and transcriptional regulation in PC. By studying somatic single nucleotide variants and germline single nucleotide polymorphisms in the cis-regulatory elements of prostate tumors, Mazrooei et al. found that these variants are specifically enriched in the cistromes of master transcriptional regulators AR, FOXA1, HOXB13, and SOX9 [100]. This implicates noncoding variation in the dysregulation of the chromatin binding activity of these factors and therefore, in prostate carcinogenesis. AR expression has further been found to be modulated through a somatically gained upstream enhancer in CRPC [101] and its binding sites have been shown to be lost or gained due to the activity of FOXA1 and HOXB13 during carcinogenesis [61]. The chromatin regions linked to altered gene regulation in PC have also been shown to be enriched in PC predisposing genetic variants. Pomerantz et al. analyzed prostate lineage-specific enhancers and promoters marked by a combination of histone modifications and PC-specific TFs AR, FOXA1, and HOXB13, and showed that these were enriched for genetic variants linked to increased PC risk, depicting an active epigenetic state [64]. In addition, they showed that prostate lineage-specific regulatory regions exhibit active epigenetic states that are predicted to increase somatic mutational burden [64]. Collectively, these studies highlight the importance of the noncoding genome in PC lineage commitment and plasticity.
4. Dysregulation of Chromatin States through Histone Modification in Prostate Cancer
The organization and structure of the chromatin is the result of the activity of various proteins interacting with DNA. The negatively charged 2 nm DNA double helix wraps around positively charged histone octamers, creating the 10 nm fiber of adjacent nucleosomes. By continuous folding and looping, this primary structure is further compacted to higher order structures to form the 3D organization of the chromatin. Both DNA replication and RNA transcription require accessibility to DNA by reversible decondensation of these chromatin structures. Chromatin compaction and decondensation at certain loci are tightly regulated and cell type-specific. This is primarily achieved through the ability of chromatin-associated proteins to write (e.g., with acetyl transferase activity), read (e.g., with bromodomains), and erase (e.g., with demethylases) the histone code.
The histone code is the set of histone modifications (methylation, acetylation, etc.) defining the state of the chromatin. For instance, a chromatin region marked by methylation in lysine 4 of the histone 3 tail (typically marked as H3K4me1) and H3K36me1 could be defined as a promoter [131,132,133][102][103][104], but additional histone markers such as H3K27 Acetylation (H2K27Ac), H3K4me3, H3K27me3, or the presence of RNAPol2 could determine whether the promoter is actually active or poised [131,132,133,134][102][103][104][105]. ChromHMM is a tool that converts the histone code to an annotated chromatin state [133,134][104][105]. The presence of histone marks H3K4me1, H3K4me3, H3K27me3, H3K27Ac, H3K36me3, and H3K9me3 are used to annotate the chromatin state based on the Roadmap Epigenomics 18-state expanded model [135][106]. For example, in this model, active transcription is marked with H3K4me3, whereas repressed chromatin is marked with H3K27me3 and H3K9me3 heterochromatin marks [135][106]
PC cells exhibit significant dysregulation of chromatin states (Figure 4). Firstly, androgen stimulation can trigger several changes in the chromatin states of PC cells. H3 acetylation has been demonstrated to accumulate at the KLK3 (encoding PSA) promoter and enhancer regions upon androgen treatment in LNCaP cells [136,137,138][107][108][109]. In addition, the presence of androgens triggers the recruitment of histone acetylases (HATs) CBP and p300 to the regulatory regions of KLK3, thus changing the chromatin state to a more active one via their acetyl transferase activity [138,139][109][110]. p300 has also been shown to protect AR from degradation in PTEN-depleted PC mouse models, in which p300 was essential for the expression of AR target genes [140][111]. Moreover, combined inhibition of p300 and CBP significantly decreases the expression of programmed death-ligand 1 (PD-L1), which increases the efficacy of immunotherapy that aims to reactivate T-cells by PD-L1 blockade [141][112].
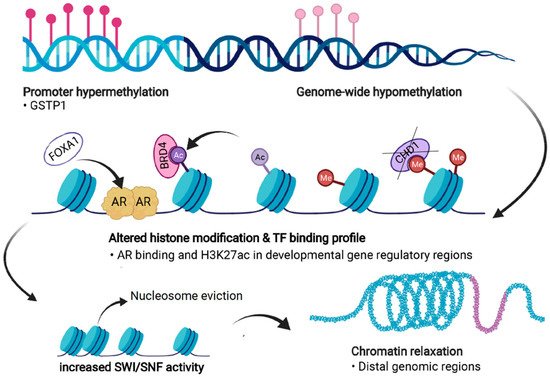
Figure 4. Forms of epigenetic and chromatin dysregulation in PC. DNA methylation is increased at specific regulatory regions, such as the GSTP1 promoter, but is generally reduced genome-wide. Aberrant histone modifications especially at distal regulatory regions, often harboring binding motifs for key transcription factors (TFs) such as the androgen receptor (AR), shift to a more active state. This also occurs due to the action of transcriptional coactivators and chromatin readers such as BRD4, leading to recruitment of these TFs to previously repressed regions. Loss of CHD1 induces chromatin rewiring, and pioneer factors such as FOXA1 are able to bind to repressed regions and recruit other TFs and histone remodeling complexes. Dysregulation of chromatin accessibility is partly due to increased activity of the SWI/SNF remodeling complex, which shifts the chromatin towards more permissive states, a process termed chromatin relaxation. Figure created with BioRender.com.
In addition to HATs, histone demethylases (JHDM2A, JMJD2C, and LSD1) have been reported to exhibit androgen stimulation-dependent recruitment to AR target genes such as KLK3, TMPRSS2, and NKX3-1, resulting in less methylated H3K9 and an active chromatin state [142,143,144][113][114][115]. Low levels of H3K9me2 have been shown to predict poor prognosis in PC patients [145][116], whereas higher IHC staining of H3K4me1 is associated with a higher risk of biochemical recurrence [146][117]. High levels of H3K4me2 are also associated with shorter time to biochemical recurrence after radical prostatectomy [147][118]. Despite active histone marks being associated with prognosis, suppressive chromatin states marked by H3K27me3 seem to be more abundant than active chromatin states marked by H3K4me3 in PC cells [148,149][119][120]. Ke et al. performed a genome-wide profiling of H3K27me3 and H3K4me3 in PC and normal epithelial cells and showed that while the number of each modification is relatively similar in both cells, the genome-wide profiles of these marks vary so that the genes marked by H3K27me3 or by both H3K27me3 and H3K4me3 are approximately 70% different [148][119]. Genes marked by H3K27me3 in PC cells are enriched with developmental functions, which was not the case for normal epithelial cells. This suggests that epigenetic reprogramming of developmental genes occurs during PC carcinogenesis. Examining genes that exhibited differential chromatin state in PC compared to normal epithelial cells, Ke et al. observed that frequent switches between H3K4me3 and H3K27me3 caused the strongest expression changes in genes, including most of the HOXA genes cluster [148][119].
Changes in chromatin states have also been associated with PC progression. The expression of the aforementioned HATs p300 and CBP was shown to be significantly higher in metastatic CRPC lesions than in local tumors [150][121]. p300 functions to stabilize a histone demethylase JMJD1A, which is upregulated in CRPC in comparison to PC, increasing JMJD1A recruitment to AR target genes and thus, facilitating the conversion of the chromatin state into an active one [151][122]. The levels of H3K4me1, H3K4me2, and H3K4me3 based on IHC increase during PC progression [146][117]. This is interesting as the binding of FOXA1 to its targets strongly depends on the distribution of H3K4me1 and H3K4me2 [93,152][93][123]. Selective accumulation of these histone marks is found at the AR-bound enhancers of M-phase cell cycle genes, such as CDK1 and UBEC2, in CRPC tissues and cell models when compared to androgen-dependent cell lines and tumor tissue samples [153][124]. Consistently analyzing global levels of H3K27me3 by IHC, Pellakuru et al. showed that the levels decrease as markers of aggressive disease increase [154][125]. This decline in H3K27me3 levels is also observed when more differentiated normal prostatic luminal epithelial cells are compared to more stem-like basal cells [154][125].
These findings suggest that overall active chromatin state increases during PC progression and that the epigenome is reprogrammed to resemble stem-like cells. Supporting this, genes repressed and marked by H3K27me3 levels in normal prostate tissue and derepressed during PC progression are developmental regulators and homeobox proteins [155,156][126][127]. Yu et al. revealed that such epigenetic reprogramming might partly originate from the aberrant function of EZH2 [155,156][126][127]. Many studies have now shown that EZH2 is overexpressed in PC and even more so in CRPC [157,158,159,160][128][129][130][131]. However, it has been shown to also have a PRC2-independent role in CRPC [161][132]. Mechanistically, EZH2 functions as a co-activator for important transcription factors such as AR upon phosphorylation [161][132], and as a part of the PRC2 complex to induce silenced chromatin states.
The combination of aberrant functions of HATs, demethylases, and EZH2 all seem to contribute to increased active chromatin states in PC progression and treatment resistance. In line with this, Pomerantz et al. showed that the profiles of active enhancers defined by H3K27ac are similar in CRPC and prostate-specific fetal tissue, implicating reactivation of the developmental transcriptional programs in late-stage disease [64].
5. Dysregulation of Chromatin Accessibility
Chromatin accessibility is facilitated by the chromatin remodeling complexes by either ejecting histones or full nucleosomes or by sliding nucleosomes to different positions in the DNA [162,163][133][134]. Several publications have linked the chromatin remodeling complex SWI/SNF to PC carcinogenesis and progression. BRG1 (also known as SMARCA4), a core subunit of the SWI/SNF complex, is required for chromatin accessibility [163][134]. Previous studies have reported the increased expression of BRG1 in primary PC [164,165,166,167][135][136][137][138]. Higher BRG1 expression has been associated with larger tumor mass and increased invasion of PC-3 cells [166][137]. Inhibition of BRG1 in PC-3 cell lines has also been demonstrated to decrease cell proliferation and induce apoptosis [168][139]. Inhibition of BRG1 in mouse xenografts increased the survival of the mice and decreased tumor growth [168][139]. Moreover, its increased expression has been associated with more malignant features [164,166,167,168][135][137][138][139]. Expression of BRG1 has a positive correlation with Gleason score, as it increases as the tumor progresses and the highest expression is observed in NEPC [164,166,167,168][135][137][138][139].
PTEN loss is associated with PC progression to CRPC [169][140] and has been demonstrated to stabilize, and thus, increase, the amount of BRG1 protein [167][138]. IHC staining of radical prostatectomy tissues demonstrated that in tumors exhibiting low PTEN expression, high BRG1 expression correlated with poor outcome [167][138]. PTEN and BRG1 were indeed shown to be synthetic lethal in a panel of PC cell lines and in a mouse model [167][138]. Open chromatin regions were reduced by approximately 60% when BRG1 was depleted in cells lacking PTEN, highlighting the importance of this SWI/SNF subunit to chromatin accessibility [167][138].
In addition to BRG1, increased expression of SWI/SNF complex subunits BAF57 and BAF155 has been described in PC [170,171,172][141][142][143]. Inhibition of BAF57 results in the inhibition of AR-dependent genes in PC cell lines [172][143]. IHC studies of primary tumors and metastatic specimens have revealed that BAF57 expression positively correlates with Gleason score [170,172][141][143]. Moreover, cell line studies with LNCaP and C4-2 (a CRPC cell line) showed that BAF57 mediates ligand-independent AR activity, thus inducing the expression of migration- and metastasis-related genes in the absence of androgens [170][141]. TMA IHC analysis of BAF155 revealed the highest staining in recurrent and metastatic prostate cancer specimens and a positive correlation with Gleason score [171][142].
Interestingly, the differential expression of certain SWI/SNF complex subunits has been linked to the progression of PC into NEPC [164][135]. For example, the RNA and protein expression of BRG1 and neuron-specific SWI/SNF subunit proteins, such as BAF53B and BAF45B, exhibited significantly higher expression in NEPC than in CRPC [164][135]. This suggests that differential chromatin remodeling is required for the progression of prostate cancer to CRPC and NEPC.
Abnormal activation of chromatin-related proteins such as chromatin readers and the SWI/SNF complex and its subunits suggests an overall increase in accessible chromatin in gene regulatory regions in CRPC. Indeed, multiple studies have shown that relaxed chromatin correlates with PC progression [19,72,145,154,173,174][19][72][116][125][144][145] and that different markers for open chromatin can function as predictors for tumor recurrence [175,176][146][147]. Moreover, Bromodomain and Extraterminal domain (BET) proteins BRD2 and BRD4, chromatin readers that recognize acetylated histones and function to promote transcription, have been shown to maintain accessible chromatin in PC [19,177,178,179][19][148][149][150] and to be overexpressed in CRPC [19]. These proteins have been demonstrated to mediate tumor growth in both CRPC cell lines and in vivo experiments with xenograft models [180,181,182,183][151][152][153][154]. In addition, BET proteins are associated with the increased expression of migration- and invasion-related genes in CRPC [184,185][155][156]. Although BRD4 can co-regulate invasion-related genes together with ERG [184][155], it seems that the main mechanism of function for these proteins is to enhance AR-mediated transcriptional regulation [180[151][152][153][154][157],181,182,183,186], highlighted by the fact that AR-negative cells are not sensitive to BET inhibitors [180][151]. AR overexpression has been associated with the chromatin accessibility maintenance activities of BET proteins BRD4 and BRD2 together with AR-regulated bromodomain-containing protein ATAD2 [19], indicating that these proteins remodel chromatin to promote AR-mediated gene regulation.
Similarly, the SWI/SNF complex has been shown to be an important transcriptional cofactor for AR activity [172,187,188,189,190][143][158][159][160][161]. Multiple studies have shown that SWI/SNF activity is critical for the AR-mediated activation of KLK3 and TMPRSS2 transcription [172,188,190][143][159][161]. In addition, SWI/SNF activity positively regulates the expression of other androgen regulated genes, such as FKBP5 and KLK2 [188][159]. Inhibition of different subunits of the SWI/SNF complex inhibits the proliferation of AR-dependent PC cells [188[159][160],189], but different subunits of the SWI/SNF complex seem to promote AR-mediated activation on different target genes [190][161]. This highlights the dynamic nature of SWI/SNF function in mediating transcriptional plasticity in response to potential PC treatments.
The importance of bromodomain activity in modifying the structure of the chromatin in PC has been reviewed previously [191][162]. Here, we add that Welti et al. recently reported that targeting p300/CBP, histone acetyltransferases with known AR transcriptional coactivator function, could be a new therapeutic strategy for CRPC, and describe a novel small-molecule inhibitor (CCS1477) of the p300/CBP conserved bromodomain [150][121].
These findings demonstrate the abnormal function of chromatin accessibility regulators in PC and in the progression to treatment resistance. These changes precede the reprogramming of TF chromatin binding, especially the binding of the AR.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Sathianathen, N.J.; Koschel, S.; Thangasamy, I.A.; Teh, J.; Alghazo, O.; Butcher, G.; Howard, H.; Kapoor, J.; Lawrentschuk, N.; Siva, S.; et al. Indirect Comparisons of Efficacy between Combination Approaches in Metastatic Hormone-Sensitive Prostate Cancer: A Systematic Review and Network Meta-Analysis. Eur. Urol. 2020, 77, 365–372.
- Heidenreich, A.; Bastian, P.J.; Bellmunt, J.; Bolla, M.; Joniau, S.; van der Kwast, T.; Mason, M.; Matveev, V.; Wiegel, T.; Zattoni, F.; et al. EAU Guidelines on Prostate Cancer. Part II: Treatment of Advanced, Relapsing, and Castration-Resistant Prostate Cancer. Eur. Urol. 2014, 65, 467–479.
- Tosco, L.; Briganti, A.; D’amico, A.V.; Eastham, J.; Eisenberger, M.; Gleave, M.; Haustermans, K.; Logothetis, C.J.; Saad, F.; Sweeney, C.; et al. Systematic Review of Systemic Therapies and Therapeutic Combinations with Local Treatments for High-Risk Localized Prostate Cancer. Eur. Urol. 2019, 75, 44–60.
- Moris, L.; Cumberbatch, M.G.; Van den Broeck, T.; Gandaglia, G.; Fossati, N.; Kelly, B.; Pal, R.; Briers, E.; Cornford, P.; De Santis, M.; et al. Benefits and Risks of Primary Treatments for High-Risk Localized and Locally Advanced Prostate Cancer: An International Multidisciplinary Systematic Review. Eur. Urol. 2020, 77, 614–627.
- Beltran, H.; Prandi, D.; Mosquera, J.M.; Benelli, M.; Puca, L.; Cyrta, J.; Marotz, C.; Giannopoulou, E.; Chakravarthi, B.V.S.K.; Varambally, S.; et al. Divergent Clonal Evolution of Castration-Resistant Neuroendocrine Prostate Cancer. Nat. Med. 2016, 22, 298–305.
- Formaggio, N.; Rubin, M.A.; Theurillat, J.-P. Loss and Revival of Androgen Receptor Signaling in Advanced Prostate Cancer. Oncogene 2021, 40, 1205–1216.
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging Mechanisms of Resistance to Androgen Receptor Inhibitors in Prostate Cancer. Nat. Rev. Cancer 2015, 15, 701–711.
- Beltran, H.; Tomlins, S.; Aparicio, A.; Arora, V.; Rickman, D.; Ayala, G.; Huang, J.; True, L.; Gleave, M.E.; Soule, H.; et al. Aggressive Variants of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2014, 20, 2846–2850.
- Handle, F.; Prekovic, S.; Helsen, C.; Van den Broeck, T.; Smeets, E.; Moris, L.; Eerlings, R.; El Kharraz, S.; Urbanucci, A.; Mills, I.G.; et al. Drivers of AR Indifferent Anti-Androgen Resistance in Prostate Cancer Cells. Sci. Rep. 2019, 9, 13786.
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017, 32, 474–489.e6.
- Crona, D.J.; Whang, Y.E. Androgen Receptor-Dependent and -Independent Mechanisms Involved in Prostate Cancer Therapy Resistance. Cancers 2017, 9, 67.
- Isikbay, M.; Otto, K.; Kregel, S.; Kach, J.; Cai, Y.; Vander Griend, D.J.; Conzen, S.D.; Szmulewitz, R.Z. Glucocorticoid Receptor Activity Contributes to Resistance to Androgen-Targeted Therapy in Prostate Cancer. Horm. Cancer 2014, 5, 72–89.
- Li, J.; Alyamani, M.; Zhang, A.; Chang, K.-H.; Berk, M.; Li, Z.; Zhu, Z.; Petro, M.; Magi-Galluzzi, C.; Taplin, M.-E.; et al. Aberrant Corticosteroid Metabolism in Tumor Cells Enables GR Takeover in Enzalutamide Resistant Prostate Cancer. eLife 2017, 6.
- Mu, P.; Zhang, Z.; Benelli, M.; Karthaus, W.R.; Hoover, E.; Chen, C.-C.; Wongvipat, J.; Ku, S.-Y.; Gao, D.; Cao, Z.; et al. SOX2 Promotes Lineage Plasticity and Antiandrogen Resistance in TP53- and RB1-Deficient Prostate Cancer. Science 2017, 355, 84–88.
- Braadland, P.R.; Ramberg, H.; Grytli, H.H.; Urbanucci, A.; Nielsen, H.K.; Guldvik, I.J.; Engedal, A.; Ketola, K.; Wang, W.; Svindland, A.; et al. The β2-Adrenergic Receptor Is a Molecular Switch for Neuroendocrine Transdifferentiation of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 2154–2168.
- Jia, L.; Shen, H.C.; Wantroba, M.; Khalid, O.; Liang, G.; Wang, Q.; Gentzschein, E.; Pinski, J.K.; Stanczyk, F.Z.; Jones, P.A.; et al. Locus-Wide Chromatin Remodeling and Enhanced Androgen Receptor-Mediated Transcription in Recurrent Prostate Tumor Cells. Mol. Cell. Biol. 2006, 26, 7331–7341.
- Tewari, A.K.; Yardimci, G.G.; Shibata, Y.; Sheffield, N.C.; Song, L.; Taylor, B.S.; Georgiev, S.G.; Coetzee, G.A.; Ohler, U.; Furey, T.S.; et al. Chromatin Accessibility Reveals Insights into Androgen Receptor Activation and Transcriptional Specificity. Genome Biol. 2012, 13, R88.
- Urbanucci, A.; Barfeld, S.J.; Kytölä, V.; Itkonen, H.M.; Coleman, I.M.; Vodák, D.; Sjöblom, L.; Sheng, X.; Tolonen, T.; Minner, S.; et al. Androgen Receptor Deregulation Drives Bromodomain-Mediated Chromatin Alterations in Prostate Cancer. Cell Rep. 2017, 19, 2045–2059.
- Brücher, B.L.D.M.; Jamall, I.S. Somatic Mutation Theory—Why It’s Wrong for Most Cancers. Cell. Physiol. Biochem. 2016, 38, 1663–1680.
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-Cancer Analysis of Whole Genomes. Nature 2020, 578, 82–93.
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome Sequencing Identifies Recurrent SPOP, FOXA1 and MED12 Mutations in Prostate Cancer. Nat. Genet. 2012, 44, 685–689.
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70.
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674.
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic Plasticity and the Hallmarks of Cancer. Science 2017, 357.
- Darwiche, N. Epigenetic Mechanisms and the Hallmarks of Cancer: An Intimate Affair. Am. J. Cancer Res. 2020, 10, 1954–1978.
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.-K.; Koche, R.P.; et al. Genome-Wide Maps of Chromatin State in Pluripotent and Lineage-Committed Cells. Nature 2007, 448, 553–560.
- Rubin, M.A.; Bristow, R.G.; Thienger, P.D.; Dive, C.; Imielinski, M. Impact of Lineage Plasticity to and from a Neuroendocrine Phenotype on Progression and Response in Prostate and Lung Cancers. Mol. Cell 2020, 80, 562–577.
- Brocks, D.; Assenov, Y.; Minner, S.; Bogatyrova, O.; Simon, R.; Koop, C.; Oakes, C.; Zucknick, M.; Lipka, D.B.; Weischenfeldt, J.; et al. Intratumor DNA Methylation Heterogeneity Reflects Clonal Evolution in Aggressive Prostate Cancer. Cell Rep. 2014, 8, 798–806.
- Fontugne, J.; Davis, K.; Palanisamy, N.; Udager, A.; Mehra, R.; McDaniel, A.S.; Siddiqui, J.; Rubin, M.A.; Mosquera, J.M.; Tomlins, S.A. Clonal Evaluation of Prostate Cancer Foci in Biopsies with Discontinuous Tumor Involvement by Dual ERG/SPINK1 Immunohistochemistry. Mod. Pathol. 2016, 29, 157–165.
- Salami, S.S.; Hovelson, D.H.; Kaplan, J.B.; Mathieu, R.; Udager, A.M.; Curci, N.E.; Lee, M.; Plouffe, K.R.; de la Vega, L.L.; Susani, M.; et al. Transcriptomic Heterogeneity in Multifocal Prostate Cancer. JCI Insight 2018, 3.
- Talos, F.; Mitrofanova, A.; Bergren, S.K.; Califano, A.; Shen, M.M. A Computational Systems Approach Identifies Synergistic Specification Genes That Facilitate Lineage Conversion to Prostate Tissue. Nat. Commun. 2017, 8, 14662.
- Xie, Q.; Liu, Y.; Cai, T.; Horton, C.; Stefanson, J.; Wang, Z.A. Dissecting Cell-Type-Specific Roles of Androgen Receptor in Prostate Homeostasis and Regeneration through Lineage Tracing. Nat. Commun. 2017, 8, 14284.
- Dutta, A.; Le Magnen, C.; Mitrofanova, A.; Ouyang, X.; Califano, A.; Abate-Shen, C. Identification of an NKX3.1-G9a-UTY Transcriptional Regulatory Network That Controls Prostate Differentiation. Science 2016, 352, 1576–1580.
- Huang, L.; Pu, Y.; Hepps, D.; Danielpour, D.; Prins, G.S. Posterior Hox Gene Expression and Differential Androgen Regulation in the Developing and Adult Rat Prostate Lobes. Endocrinology 2007, 148, 1235–1245.
- Javed, S.; Langley, S.E.M. Importance of HOX Genes in Normal Prostate Gland Formation, Prostate Cancer Development and Its Early Detection. BJU Int. 2014, 113, 535–540.
- Guo, W.; Li, L.; He, J.; Liu, Z.; Han, M.; Li, F.; Xia, X.; Zhang, X.; Zhu, Y.; Wei, Y.; et al. Single-Cell Transcriptomics Identifies a Distinct Luminal Progenitor Cell Type in Distal Prostate Invagination Tips. Nat. Genet. 2020, 52, 908–918.
- Isaacs, J.T. Control of cell proliferation and cell death in the normal and neoplastic prostate: A stem cell model. In Benign Prostatic Hyperplasia; Rogers, C.H., Coffey, D.S., Cunha, G., Grayhack, J.T., Hinman, F., Jr., Horton, R., Eds.; Report No. NIH 87-2881; US Department of Health and Human Services: Washington, DC, USA, 1987; pp. 85–94.
- Tsujimura, A.; Koikawa, Y.; Salm, S.; Takao, T.; Coetzee, S.; Moscatelli, D.; Shapiro, E.; Lepor, H.; Sun, T.-T.; Wilson, E.L. Proximal Location of Mouse Prostate Epithelial Stem Cells: A Model of Prostatic Homeostasis. J. Cell Biol. 2002, 157, 1257–1265.
- Henry, G.H.; Malewska, A.; Joseph, D.B.; Malladi, V.S.; Lee, J.; Torrealba, J.; Mauck, R.J.; Gahan, J.C.; Raj, G.V.; Roehrborn, C.G.; et al. A Cellular Anatomy of the Normal Adult Human Prostate and Prostatic Urethra. Cell Rep. 2018, 25, 3530–3542.e5.
- Karthaus, W.R.; Hofree, M.; Choi, D.; Linton, E.L.; Turkekul, M.; Bejnood, A.; Carver, B.; Gopalan, A.; Abida, W.; Laudone, V.; et al. Regenerative Potential of Prostate Luminal Cells Revealed by Single-Cell Analysis. Science 2020, 368, 497–505.
- Montoro, D.T.; Haber, A.L.; Biton, M.; Vinarsky, V.; Lin, B.; Birket, S.E.; Yuan, F.; Chen, S.; Leung, H.M.; Villoria, J.; et al. A Revised Airway Epithelial Hierarchy Includes CFTR-Expressing Ionocytes. Nature 2018, 560, 319–324.
- Maitland, N.J.; Collins, A.T. Prostate Cancer Stem Cells: A New Target for Therapy. J. Clin. Oncol. 2008, 26, 2862–2870.
- Hudson, D.L.; Guy, A.T.; Fry, P.; O’Hare, M.J.; Watt, F.M.; Masters, J.R. Epithelial Cell Differentiation Pathways in the Human Prostate: Identification of Intermediate Phenotypes by Keratin Expression. J. Histochem. Cytochem. 2001, 49, 271–278.
- Wang, Z.A.; Mitrofanova, A.; Bergren, S.K.; Abate-Shen, C.; Cardiff, R.D.; Califano, A.; Shen, M.M. Lineage Analysis of Basal Epithelial Cells Reveals Their Unexpected Plasticity and Supports a Cell-of-Origin Model for Prostate Cancer Heterogeneity. Nat. Cell Biol. 2013, 15, 274–283.
- Wang, X.; Julio, M.K.; Economides, K.D.; Walker, D.; Yu, H.; Vivienne Halili, M.; Hu, Y.-P.; Price, S.M.; Abate-Shen, C.; Shen, M.M. A Luminal Epithelial Stem Cell That Is a Cell of Origin for Prostate Cancer. Nature 2009, 461, 495–500.
- Goldstein, A.S.; Huang, J.; Guo, C.; Garraway, I.P.; Witte, O.N. Identification of a Cell of Origin for Human Prostate Cancer. Science 2010, 329, 568–571.
- Choi, N.; Zhang, B.; Zhang, L.; Ittmann, M.; Xin, L. Adult Murine Prostate Basal and Luminal Cells Are Self-Sustained Lineages That Can Both Serve as Targets for Prostate Cancer Initiation. Cancer Cell 2012, 21, 253–265.
- Wang, Z.A.; Toivanen, R.; Bergren, S.K.; Chambon, P.; Shen, M.M. Luminal Cells Are Favored as the Cell of Origin for Prostate Cancer. Cell Rep. 2014, 8, 1339–1346.
- Lee, S.H.; Shen, M.M. Cell Types of Origin for Prostate Cancer. Curr. Opin. Cell Biol. 2015, 37, 35–41.
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The Inflammatory Microenvironment and Microbiome in Prostate Cancer Development. Nat. Rev. Urol. 2018, 15, 11–24.
- Guo, H.; Ci, X.; Ahmed, M.; Hua, J.T.; Soares, F.; Lin, D.; Puca, L.; Vosoughi, A.; Xue, H.; Li, E.; et al. ONECUT2 Is a Driver of Neuroendocrine Prostate Cancer. Nat. Commun. 2019, 10, 278.
- Giunchi, F.; Fiorentino, M.; Loda, M. The Metabolic Landscape of Prostate Cancer. Eur. Urol. Oncol. 2019, 2, 28–36.
- Corbin, J.M.; Ruiz-Echevarría, M.J. One-Carbon Metabolism in Prostate Cancer: The Role of Androgen Signaling. Int. J. Mol. Sci. 2016, 17, 1208.
- Gurel, B.; Iwata, T.; Koh, C.M.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear MYC Protein Overexpression Is an Early Alteration in Human Prostate Carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167.
- Civenni, G.; Malek, A.; Albino, D.; Garcia-Escudero, R.; Napoli, S.; Di Marco, S.; Pinton, S.; Sarti, M.; Carbone, G.M.; Catapano, C.V. RNAi-Mediated Silencing of Myc Transcription Inhibits Stem-like Cell Maintenance and Tumorigenicity in Prostate Cancer. Cancer Res. 2013, 73, 6816–6827.
- Vander Griend, D.J.; Litvinov, I.V.; Isaacs, J.T. Conversion of Androgen Receptor Signaling from a Growth Suppressor in Normal Prostate Epithelial Cells to an Oncogene in Prostate Cancer Cells Involves a Gain of Function in c-Myc Regulation. Int. J. Biol. Sci. 2014, 10, 627–642.
- Antony, L.; van der Schoor, F.; Dalrymple, S.L.; Isaacs, J.T. Androgen Receptor (AR) Suppresses Normal Human Prostate Epithelial Cell Proliferation via AR/β-catenin/TCF-4 Complex Inhibition of c-MYC Transcription. Prostate 2014, 74, 1118–1131.
- Maitland, N.J.; Collins, A. A Tumour Stem Cell Hypothesis for the Origins of Prostate Cancer. BJU Int. 2005, 96, 1219–1223.
- Song, H.; Weinstein, H.N.W.; Allegakoen, P.; Wadsworth, M.H.; Xie, J.; Yang, H.; Feng, F.Y.; Carroll, P.R.; Wang, B.; Cooperberg, M.R.; et al. Single-Cell Analysis of Human Primary Prostate Cancer Reveals the Heterogeneity of Tumor-Associated Epithelial Cell States. bioRxiv 2020.
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The Androgen Receptor Cistrome Is Extensively Reprogrammed in Human Prostate Tumorigenesis. Nat. Genet. 2015, 47, 1346–1351.
- Grbesa, I.; Augello, M.A.; Liu, D.; McNally, D.R.; Gaffney, C.D. SPOP Mutation Confers Sensitivity to AR-Targeted Therapy in Prostate Cancer by Reshaping the Androgen-Driven Chromatin Landscape. bioRxiv 2021.
- Sharma, N.L.; Massie, C.E.; Ramos-Montoya, A.; Zecchini, V.; Scott, H.E.; Lamb, A.D.; MacArthur, S.; Stark, R.; Warren, A.Y.; Mills, I.G.; et al. The Androgen Receptor Induces a Distinct Transcriptional Program in Castration-Resistant Prostate Cancer in Man. Cancer Cell 2013, 23, 35–47.
- Pomerantz, M.M.; Qiu, X.; Zhu, Y.; Takeda, D.Y.; Pan, W.; Baca, S.C.; Gusev, A.; Korthauer, K.D.; Severson, T.M.; Ha, G.; et al. Prostate Cancer Reactivates Developmental Epigenomic Programs during Metastatic Progression. Nat. Genet. 2020, 52, 790–799.
- Kwon, O.-J.; Zhang, L.; Jia, D.; Zhou, Z.; Li, Z.; Haffner, M.; Lee, J.K.; True, L.; Morrissey, C.; Xin, L. De Novo Induction of Lineage Plasticity from Human Prostate Luminal Epithelial Cells by Activated AKT1 and c-Myc. Oncogene 2020, 39, 7142–7151.
- Carceles-Cordon, M.; Kelly, W.K.; Gomella, L.; Knudsen, K.E.; Rodriguez-Bravo, V.; Domingo-Domenech, J. Cellular Rewiring in Lethal Prostate Cancer: The Architect of Drug Resistance. Nat. Rev. Urol. 2020, 17, 292–307.
- Linja, M.J.; Savinainen, K.J.; Saramäki, O.R.; Tammela, T.L.; Vessella, R.L.; Visakorpi, T. Amplification and Overexpression of Androgen Receptor Gene in Hormone-Refractory Prostate Cancer. Cancer Res. 2001, 61, 3550–3555.
- Zhang, Z.; Zhou, C.; Li, X.; Barnes, S.D.; Deng, S.; Hoover, E.; Chen, C.-C.; Lee, Y.S.; Zhang, Y.; Wang, C.; et al. Loss of CHD1 Promotes Heterogeneous Mechanisms of Resistance to AR-Targeted Therapy via Chromatin Dysregulation. Cancer Cell 2020, 37, 584–598.e11.
- Brennen, W.N.; Nathaniel Brennen, W.; Zhu, Y.; Coleman, I.M.; Dalrymple, S.L.; Antony, L.; Patel, R.A.; Hanratty, B.; Chikarmane, R.; Meeker, A.K.; et al. Resistance to Androgen Receptor Signaling Inhibition Does Not Necessitate Development of Neuroendocrine Prostate Cancer. JCI Insight 2021, 6, e146827.
- Nyquist, M.D.; Corella, A.; Coleman, I.; De Sarkar, N.; Kaipainen, A.; Ha, G.; Gulati, R.; Ang, L.; Chatterjee, P.; Lucas, J.; et al. Combined TP53 and RB1 Loss Promotes Prostate Cancer Resistance to a Spectrum of Therapeutics and Confers Vulnerability to Replication Stress. Cell Rep. 2020, 31, 107669.
- He, M.X.; Cuoco, M.S.; Crowdis, J.; Bosma-Moody, A.; Zhang, Z.; Bi, K.; Kanodia, A.; Su, M.-J.; Ku, S.-Y.; Garcia, M.M.; et al. Transcriptional Mediators of Treatment Resistance in Lethal Prostate Cancer. Nat. Med. 2021, 27, 426–433.
- Uusi-Mäkelä, J.; Afyounian, E.; Tabaro, F.; Häkkinen, T.; Lussana, A.; Shcherban, A.; Annala, M.; Nurminen, R.; Kivinummi, K.; Tammela, T.L.J.; et al. Chromatin Accessibility Analysis Uncovers Regulatory Element Landscape in Prostate Cancer Progression. bioRxiv 2020.
- Ku, S.Y.; Rosario, S.; Wang, Y.; Mu, P.; Seshadri, M.; Goodrich, Z.W.; Goodrich, M.M.; Labbé, D.P.; Gomez, E.C.; Wang, J.; et al. Rb1 and Trp53 Cooperate to Suppress Prostate Cancer Lineage Plasticity, Metastasis, and Antiandrogen Resistance. Science 2017, 355, 78–83.
- Mandigo, A.C.; Yuan, W.; Xu, K.; Gallagher, P.; Pang, A.; Guan, Y.F.; Shafi, A.A.; Thangavel, C.; Sheehan, B.; Bogdan, D.; et al. RB/E2F1 as a Master Regulator of Cancer Cell Metabolism in Advanced Disease. Cancer Discov. 2021.
- Kaarijärvi, R.; Kaljunen, H.; Ketola, K. Molecular and Functional Links between Neurodevelopmental Processes and Treatment-Induced Neuroendocrine Plasticity in Prostate Cancer Progression. Cancers 2021, 13, 692.
- Park, J.W.; Lee, J.K.; Sheu, K.M.; Wang, L.; Balanis, N.G.; Nguyen, K.; Smith, B.A.; Cheng, C.; Tsai, B.L.; Cheng, D.; et al. Reprogramming Normal Human Epithelial Tissues to a Common, Lethal Neuroendocrine Cancer Lineage. Science 2018, 362, 91–95.
- Baca, S.C.; Takeda, D.Y.; Seo, J.-H.; Hwang, J.; Ku, S.Y.; Arafeh, R.; Arnoff, T.; Agarwal, S.; Bell, C.; O’Connor, E.; et al. Reprogramming of the FOXA1 Cistrome in Treatment-Emergent Neuroendocrine Prostate Cancer. Nat. Commun. 2021, 12, 1979.
- Flores-Morales, A.; Bergmann, T.B.; Lavallee, C.; Batth, T.S.; Lin, D.; Lerdrup, M.; Friis, S.; Bartels, A.; Kristensen, G.; Krzyzanowska, A.; et al. Proteogenomic Characterization of Patient-Derived Xenografts Highlights the Role of REST in Neuroendocrine Differentiation of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2019, 25, 595–608.
- Svensson, C.; Ceder, J.; Iglesias-Gato, D.; Chuan, Y.-C.; Pang, S.T.; Bjartell, A.; Martinez, R.M.; Bott, L.; Helczynski, L.; Ulmert, D.; et al. REST Mediates Androgen Receptor Actions on Gene Repression and Predicts Early Recurrence of Prostate Cancer. Nucleic Acids Res. 2014, 42, 999–1015.
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53.
- Ge, R.; Wang, Z.; Montironi, R.; Jiang, Z.; Cheng, M.; Santoni, M.; Huang, K.; Massari, F.; Lu, X.; Cimadamore, A.; et al. Epigenetic Modulations and Lineage Plasticity in Advanced Prostate Cancer. Ann. Oncol. 2020, 31, 470–479.
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc–mediated Epigenetic Reprogramming Drives Lineage Plasticity in Advanced Prostate Cancer. J. Clin. Investig. 2019, 129, 3924–3940.
- Chatterjee, A.; Rodger, E.J.; Eccles, M.R. Epigenetic Drivers of Tumourigenesis and Cancer Metastasis. Semin. Cancer Biol. 2018, 51, 149–159.
- Ruggero, K.; Farran-Matas, S.; Martinez-Tebar, A.; Aytes, A. Epigenetic Regulation in Prostate Cancer Progression. Curr. Mol. Biol. Rep. 2018, 4, 101–115.
- Heinäniemi, M.; Nykter, M.; Kramer, R.; Wienecke-Baldacchino, A.; Sinkkonen, L.; Zhou, J.X.; Kreisberg, R.; Kauffman, S.A.; Huang, S.; Shmulevich, I. Gene-Pair Expression Signatures Reveal Lineage Control. Nat. Methods 2013, 10, 577–583.
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabé, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International Network of Cancer Genome Projects. Nature 2010, 464, 993–998.
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228.
- Grasso, C.S.; Wu, Y.-M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243.
- Ylipää, A.; Kivinummi, K.; Kohvakka, A.; Annala, M.; Latonen, L.; Scaravilli, M.; Kartasalo, K.; Leppänen, S.-P.; Karakurt, S.; Seppälä, J.; et al. Transcriptome Sequencing Reveals PCAT5 as a Novel ERG-Regulated Long Noncoding RNA in Prostate Cancer. Cancer Res. 2015, 75, 4026–4031.
- Han, Y.-C.; Zheng, Z.-L.; Zuo, Z.-H.; Yu, Y.P.; Chen, R.; Tseng, G.C.; Nelson, J.B.; Luo, J.-H. Metallothionein 1 H Tumour Suppressor Activity in Prostate Cancer Is Mediated by Euchromatin Methyltransferase 1. J. Pathol. 2013, 230, 184–193.
- Augello, M.A.; Hickey, T.E.; Knudsen, K.E. FOXA1: Master of Steroid Receptor Function in Cancer. EMBO J. 2011, 30, 3885–3894.
- Teng, M.; Zhou, S.; Cai, C.; Lupien, M.; He, H.H. Pioneer of Prostate Cancer: Past, Present and the Future of FOXA1. Protein Cell 2021, 12, 29–38.
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Wang, Q.; Zhang, Y.; Li, W.; Carroll, J.S.; Liu, X.S.; Brown, M. FoxA1 Translates Epigenetic Signatures into Enhancer-Driven Lineage-Specific Transcription. Cell 2008, 132, 958–970.
- Heinlein, C.A.; Chang, C. Androgen Receptor in Prostate Cancer. Endocr. Rev. 2004, 25, 276–308.
- Rao, R.C.; Dou, Y. Hijacked in Cancer: The KMT2 (MLL) Family of Methyltransferases. Nat. Rev. Cancer 2015, 15, 334–346.
- Stokes, D.G.; Tartof, K.D.; Perry, R.P. CHD1 Is Concentrated in Interbands and Puffed Regions of Drosophila Polytene Chromosomes. Proc. Natl. Acad. Sci. USA 1996, 93, 7137–7142.
- Boysen, G.; Rodrigues, D.N.; Rescigno, P.; Seed, G.; Dolling, D.; Riisnaes, R.; Crespo, M.; Zafeiriou, Z.; Sumanasuriya, S.; Bianchini, D.; et al. SPOP-Mutated/CHD1-Deleted Lethal Prostate Cancer and Abiraterone Sensitivity. Clin. Cancer Res. 2018, 24, 5585–5593.
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, V.; et al. CHD1 Loss Sensitizes Prostate Cancer to DNA Damaging Therapy by Promoting Error-Prone Double-Strand Break Repair. Ann. Oncol. 2017, 28, 1495–1507.
- Aparicio, A.M.; Shen, L.; Tapia, E.L.N.; Lu, J.-F.; Chen, H.-C.; Zhang, J.; Wu, G.; Wang, X.; Troncoso, P.; Corn, P.; et al. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530.
- Mazrooei, P.; Kron, K.J.; Zhu, Y.; Zhou, S.; Grillo, G.; Mehdi, T.; Ahmed, M.; Severson, T.M.; Guilhamon, P.; Armstrong, N.S.; et al. Cistrome Partitioning Reveals Convergence of Somatic Mutations and Risk Variants on Master Transcription Regulators in Primary Prostate Tumors. Cancer Cell 2019, 36, 674–689.e6.
- Takeda, D.Y.; Spisák, S.; Seo, J.-H.; Bell, C.; O’Connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szállási, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e13.
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, Erasing and Reading Histone Lysine Methylations. Exp. Mol. Med. 2017, 49, e324.
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395.
- Ernst, J.; Kellis, M. ChromHMM: Automating Chromatin-State Discovery and Characterization. Nat. Methods 2012, 9, 215–216.
- Ernst, J.; Kellis, M. Chromatin-State Discovery and Genome Annotation with ChromHMM. Nat. Protoc. 2017, 12, 2478–2492.
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative Analysis of 111 Reference Human Epigenomes. Nature 2015, 518, 317–330.
- Chen, Z.; Wang, L.; Wang, Q.; Li, W. Histone Modifications and Chromatin Organization in Prostate Cancer. Epigenomics 2010, 2, 551–560.
- Kang, Z.; Jänne, O.A.; Palvimo, J.J. Coregulator Recruitment and Histone Modifications in Transcriptional Regulation by the Androgen Receptor. Mol. Endocrinol. 2004, 18, 2633–2648.
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and Temporal Recruitment of Androgen Receptor and Its Coactivators Involves Chromosomal Looping and Polymerase Tracking. Mol. Cell 2005, 19, 631–642.
- Shang, Y.; Myers, M.; Brown, M. Formation of the Androgen Receptor Transcription Complex. Mol. Cell 2002, 9, 601–610.
- Zhong, J.; Ding, L.; Bohrer, L.R.; Pan, Y.; Liu, P.; Zhang, J.; Sebo, T.J.; Karnes, R.J.; Tindall, D.J.; van Deursen, J.; et al. p300 Acetyltransferase Regulates Androgen Receptor Degradation and PTEN-Deficient Prostate Tumorigenesis. Cancer Res. 2014, 74, 1870–1880.
- Liu, J.; He, D.; Cheng, L.; Huang, C.; Zhang, Y.; Rao, X.; Kong, Y.; Li, C.; Zhang, Z.; Liu, J.; et al. p300/CBP Inhibition Enhances the Efficacy of Programmed Death-Ligand 1 Blockade Treatment in Prostate Cancer. Oncogene 2020, 39, 3939–3951.
- Yamane, K.; Toumazou, C.; Tsukada, Y.-I.; Erdjument-Bromage, H.; Tempst, P.; Wong, J.; Zhang, Y. JHDM2A, a JmjC-Containing H3K9 Demethylase, Facilitates Transcription Activation by Androgen Receptor. Cell 2006, 125, 483–495.
- Wissmann, M.; Yin, N.; Müller, J.M.; Greschik, H.; Fodor, B.D.; Jenuwein, T.; Vogler, C.; Schneider, R.; Günther, T.; Buettner, R.; et al. Cooperative Demethylation by JMJD2C and LSD1 Promotes Androgen Receptor-Dependent Gene Expression. Nat. Cell Biol. 2007, 9, 347–353.
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 Demethylates Repressive Histone Marks to Promote Androgen-Receptor-Dependent Transcription. Nature 2005, 437, 436–439.
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global Levels of Histone Modifications Predict Prognosis in Different Cancers. Am. J. Pathol. 2009, 174, 1619–1628.
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Rogenhofer, S.; Heukamp, L.C.; Gütgemann, I.; Walter, B.; Hofstädter, F.; Büttner, R.; Müller, S.C.; et al. Global Levels of Histone Modifications Predict Prostate Cancer Recurrence. Prostate 2010, 70, 61–69.
- Bianco-Miotto, T.; Chiam, K.; Buchanan, G.; Jindal, S.; Day, T.K.; Thomas, M.; Pickering, M.A.; O’Loughlin, M.A.; Ryan, N.K.; Raymond, W.A.; et al. Global Levels of Specific Histone Modifications and an Epigenetic Gene Signature Predict Prostate Cancer Progression and Development. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2611–2622.
- Ke, X.-S.; Qu, Y.; Rostad, K.; Li, W.-C.; Lin, B.; Halvorsen, O.J.; Haukaas, S.A.; Jonassen, I.; Petersen, K.; Goldfinger, N.; et al. Genome-Wide Profiling of Histone h3 Lysine 4 and Lysine 27 Trimethylation Reveals an Epigenetic Signature in Prostate Carcinogenesis. PLoS ONE 2009, 4, e4687.
- Gal-Yam, E.N.; Egger, G.; Iniguez, L.; Holster, H.; Einarsson, S.; Zhang, X.; Lin, J.C.; Liang, G.; Jones, P.A.; Tanay, A. Frequent Switching of Polycomb Repressive Marks and DNA Hypermethylation in the PC3 Prostate Cancer Cell Line. Proc. Natl. Acad. Sci. USA 2008, 105, 12979–12984.
- Welti, J.; Sharp, A.; Brooks, N.; Yuan, W.; McNair, C.; Chand, S.N.; Pal, A.; Figueiredo, I.; Riisnaes, R.; Gurel, B.; et al. Targeting the p300/CBP Axis in Lethal Prostate Cancer. Cancer Discov. 2021.
- Xu, S.; Fan, L.; Jeon, H.-Y.; Zhang, F.; Cui, X.; Mickle, M.B.; Peng, G.; Hussain, A.; Fazli, L.; Gleave, M.E.; et al. p300-Mediated Acetylation of Histone Demethylase JMJD1A Prevents Its Degradation by Ubiquitin Ligase STUB1 and Enhances Its Activity in Prostate Cancer. Cancer Res. 2020, 80, 3074–3087.
- Sahu, B.; Laakso, M.; Ovaska, K.; Mirtti, T.; Lundin, J.; Rannikko, A.; Sankila, A.; Turunen, J.-P.; Lundin, M.; Konsti, J.; et al. Dual Role of FoxA1 in Androgen Receptor Binding to Chromatin, Androgen Signalling and Prostate Cancer. EMBO J. 2011, 30, 3962–3976.
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen Receptor Regulates a Distinct Transcription Program in Androgen-Independent Prostate Cancer. Cell 2009, 138, 245–256.
- Pellakuru, L.G.; Iwata, T.; Gurel, B.; Schultz, D.; Hicks, J.; Bethel, C.; Yegnasubramanian, S.; De Marzo, A.M. Global Levels of H3K27me3 Track with Differentiation in Vivo and Are Deregulated by MYC in Prostate Cancer. Am. J. Pathol. 2012, 181, 560–569.
- Yu, J.; Cao, Q.; Mehra, R.; Laxman, B.; Yu, J.; Tomlins, S.A.; Creighton, C.J.; Dhanasekaran, S.M.; Shen, R.; Chen, G.; et al. Integrative Genomics Analysis Reveals Silencing of Beta-Adrenergic Signaling by Polycomb in Prostate Cancer. Cancer Cell 2007, 12, 419–431.
- Yu, J.; Yu, J.; Rhodes, D.R.; Tomlins, S.A.; Cao, X.; Chen, G.; Mehra, R.; Wang, X.; Ghosh, D.; Shah, R.B.; et al. A Polycomb Repression Signature in Metastatic Prostate Cancer Predicts Cancer Outcome. Cancer Res. 2007, 67, 10657–10663.
- Bryant, R.J.; Cross, N.A.; Eaton, C.L.; Hamdy, F.C.; Cunliffe, V.T. EZH2 Promotes Proliferation and Invasiveness of Prostate Cancer Cells. Prostate 2007, 67, 547–556.
- Dundr, P.; Bártů, M.; Hojný, J.; Michálková, R.; Hájková, N.; Stružinská, I.; Krkavcová, E.; Hadravský, L.; Kleissnerová, L.; Kopejsková, J.; et al. HNF1B, EZH2 and ECI2 in Prostate Carcinoma. Molecular, Immunohistochemical and Clinico-Pathological Study. Sci. Rep. 2020, 10, 14365.
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Otte, A.P.; et al. The Polycomb Group Protein EZH2 Is Involved in Progression of Prostate Cancer. Nature 2002, 419, 624–629.
- Melling, N.; Thomsen, E.; Tsourlakis, M.C.; Kluth, M.; Hube-Magg, C.; Minner, S.; Koop, C.; Graefen, M.; Heinzer, H.; Wittmer, C.; et al. Overexpression of Enhancer of Zeste Homolog 2 (EZH2) Characterizes an Aggressive Subset of Prostate Cancers and Predicts Patient Prognosis Independently from Pre- and Postoperatively Assessed Clinicopathological Parameters. Carcinogenesis 2015, 36, 1333–1340.
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 Oncogenic Activity in Castration-Resistant Prostate Cancer Cells Is Polycomb-Independent. Science 2012, 338, 1465–1469.
- Clapier, C.R.; Iwasa, J.; Cairns, B.R.; Peterson, C.L. Mechanisms of Action and Regulation of ATP-Dependent Chromatin-Remodelling Complexes. Nat. Rev. Mol. Cell Biol. 2017, 18, 407–422.
- Hargreaves, D.C. Chromatin Openness Requires Continuous SWI/SNF Activity. Nat. Genet. 2021, 53, 263–264.
- Cyrta, J.; Augspach, A.; De Filippo, M.R.; Prandi, D.; Thienger, P.; Benelli, M.; Cooley, V.; Bareja, R.; Wilkes, D.; Chae, S.-S.; et al. Role of Specialized Composition of SWI/SNF Complexes in Prostate Cancer Lineage Plasticity. Nat. Commun. 2020, 11, 5549.
- Giles, K.A.; Gould, C.M.; Achinger-Kawecka, J.; Page, S.G.; Kafer, G.R.; Rogers, S.; Luu, P.-L.; Cesare, A.J.; Clark, S.J.; Taberlay, P.C. BRG1 Knockdown Inhibits Proliferation through Multiple Cellular Pathways in Prostate Cancer. Clin. Epigenet. 2021, 13, 37.
- Sun, A.; Tawfik, O.; Gayed, B.; Thrasher, J.B.; Hoestje, S.; Li, C.; Li, B. Aberrant Expression of SWI/SNF Catalytic Subunits BRG1/BRM Is Associated with Tumor Development and Increased Invasiveness in Prostate Cancers. Prostate 2007, 67, 203–213.
- Ding, Y.; Li, N.; Dong, B.; Guo, W.; Wei, H.; Chen, Q.; Yuan, H.; Han, Y.; Chang, H.; Kan, S.; et al. Chromatin Remodeling ATPase BRG1 and PTEN Are Synthetic Lethal in Prostate Cancer. J. Clin. Investig. 2019, 129, 759–773.
- Muthuswami, R.; Bailey, L.; Rakesh, R.; Imbalzano, A.N.; Nickerson, J.A.; Hockensmith, J.W. BRG1 Is a Prognostic Indicator and a Potential Therapeutic Target for Prostate Cancer. J. Cell. Physiol. 2019.
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical Implications of PTEN Loss in Prostate Cancer. Nat. Rev. Urol. 2018, 15, 222–234.
- Balasubramaniam, S.; Comstock, C.E.S.; Ertel, A.; Jeong, K.W.; Stallcup, M.R.; Addya, S.; McCue, P.A.; Ostrander, W.F., Jr.; Augello, M.A.; Knudsen, K.E. Aberrant BAF57 Signaling Facilitates Prometastatic Phenotypes. Clin. Cancer Res. 2013, 19, 2657–2667.
- Heebøll, S.; Borre, M.; Ottosen, P.D.; Andersen, C.L.; Mansilla, F.; Dyrskjøt, L.; Orntoft, T.F.; Tørring, N. SMARCC1 Expression Is Upregulated in Prostate Cancer and Positively Correlated with Tumour Recurrence and Dedifferentiation. Histol. Histopathol. 2008, 23, 1069–1076.
- Link, K.A.; Balasubramaniam, S.; Sharma, A.; Comstock, C.E.S.; Godoy-Tundidor, S.; Powers, N.; Cao, K.H.; Haelens, A.; Claessens, F.; Revelo, M.P.; et al. Targeting the BAF57 SWI/SNF Subunit in Prostate Cancer: A Novel Platform to Control Androgen Receptor Activity. Cancer Res. 2008, 68, 4551–4558.
- Stelloo, S.; Nevedomskaya, E.; van der Poel, H.G.; de Jong, J.; van Leenders, G.J.L.H.; Jenster, G.; Wessels, L.F.A.; Bergman, A.M.; Zwart, W. Androgen Receptor Profiling Predicts Prostate Cancer Outcome. EMBO Mol. Med. 2015, 7, 1450–1464.
- Xu, Q.; Liu, X.; Zhu, S.; Hu, X.; Niu, H.; Zhang, X.; Zhu, D.; Nesa, E.U.; Tian, K.; Yuan, H. Hyper-Acetylation Contributes to the Sensitivity of Chemo-Resistant Prostate Cancer Cells to Histone Deacetylase Inhibitor Trichostatin A. J. Cell. Mol. Med. 2018, 22, 1909–1922.
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global Histone Modification Patterns Predict Risk of Prostate Cancer Recurrence. Nature 2005, 435, 1262–1266.
- Zhou, L.-X.; Li, T.; Huang, Y.-R.; Sha, J.-J.; Sun, P.; Li, D. Application of Histone Modification in the Risk Prediction of the Biochemical Recurrence after Radical Prostatectomy. Asian J. Androl. 2010, 12, 171–179.
- Devaiah, B.N.; Case-Borden, C.; Gegonne, A.; Hsu, C.H.; Chen, Q.; Meerzaman, D.; Dey, A.; Ozato, K.; Singer, D.S. BRD4 Is a Histone Acetyltransferase That Evicts Nucleosomes from Chromatin. Nat. Struct. Mol. Biol. 2016, 23, 540–548.
- Surface, L.E.; Fields, P.A.; Subramanian, V.; Behmer, R.; Udeshi, N.; Peach, S.E.; Carr, S.A.; Jaffe, J.D.; Boyer, L.A. H2A.Z.1 Monoubiquitylation Antagonizes BRD2 to Maintain Poised Chromatin in ESCs. Cell Rep. 2016, 14, 1142–1155.
- Stathis, A.; Bertoni, F. BET Proteins as Targets for Anticancer Treatment. Cancer Discov. 2018, 8, 24–36.
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic Targeting of BET Bromodomain Proteins in Castration-Resistant Prostate Cancer. Nature 2014, 510, 278–282.
- Asangani, I.A.; Wilder-Romans, K.; Dommeti, V.L.; Krishnamurthy, P.M.; Apel, I.J.; Escara-Wilke, J.; Plymate, S.R.; Navone, N.M.; Wang, S.; Feng, F.Y.; et al. BET Bromodomain Inhibitors Enhance Efficacy and Disrupt Resistance to AR Antagonists in the Treatment of Prostate Cancer. Mol. Cancer Res. 2016, 14, 324–331.
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.K.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-Induced BET Protein Degradation as a Therapy for Castration-Resistant Prostate Cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129.
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.C.; Korenchuk, S. Inhibition of BET Bromodomain Proteins as a Therapeutic Approach in Prostate Cancer. Oncotarget 2013, 4, 2419–2429.
- Blee, A.M.; Liu, S.; Wang, L.; Huang, H. BET Bromodomain-Mediated Interaction between ERG and BRD4 Promotes Prostate Cancer Cell Invasion. Oncotarget 2016, 7, 38319–38332.
- Shafran, J.S.; Andrieu, G.P.; Györffy, B.; Denis, G.V. BRD4 Regulates Metastatic Potential of Castration-Resistant Prostate Cancer through AHNAK. Mol. Cancer Res. 2019, 17.
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R. Selective Inhibition of the BD2 Bromodomain of BET Proteins in Prostate Cancer. Nature 2020, 578, 306–310.
- Huang, Z.-Q.; Li, J.; Sachs, L.M.; Cole, P.A.; Wong, J. A Role for Cofactor-Cofactor and Cofactor-Histone Interactions in Targeting p300, SWI/SNF and Mediator for Transcription. EMBO J. 2003, 22, 2146–2155.
- Jin, M.L.; Kim, Y.W.; Jeong, K.W. BAF53A Regulates Androgen Receptor-Mediated Gene Expression and Proliferation in LNCaP Cells. Biochem. Biophys. Res. Commun. 2018, 505, 618–623.
- Link, K.A.; Burd, C.J.; Williams, E.; Marshall, T.; Rosson, G.; Henry, E.; Weissman, B.; Knudsen, K.E. BAF57 Governs Androgen Receptor Action and Androgen-Dependent Proliferation through SWI/SNF. Mol. Cell. Biol. 2005, 25, 2200–2215.
- Marshall, T.W.; Link, K.A.; Petre-Draviam, C.E.; Knudsen, K.E. Differential Requirement of SWI/SNF for Androgen Receptor Activity. J. Biol. Chem. 2003, 278, 30605–30613.
- Urbanucci, A.; Mills, I.G. Bromodomain-Containing Proteins in Prostate Cancer. Mol. Cell. Endocrinol. 2018, 462, 31–40.
More