Ischemic stroke is the second cause of mortality and the first cause of long-term disability constituting a serious socioeconomic burden worldwide. Approved treatments include thrombectomy and rtPA intravenous administration, which, despite their efficacy in some cases, are not suitable for a great proportion of patients. Glial cell-related therapies are progressively overcoming inefficient neuron-centered approaches in the preclinical phase. Exploiting the ability of microglia to naturally switch between detrimental and protective phenotypes represents a promising therapeutic treatment, in a similar way to what happens with astrocytes. However, the duality present in many of the roles of these cells upon ischemia poses a notorious difficulty in disentangling the precise pathways to target. Still, promoting M2/A2 microglia/astrocyte protective phenotypes and inhibiting M1/A1 neurotoxic profiles is globally rendering promising results in different in vivo models of stroke.
- ischemic stroke
- glia
- neuroprotection
- microglia
- astrocytes
- oligodendrocytes
- therapy
1. Introduction
2. Microglia
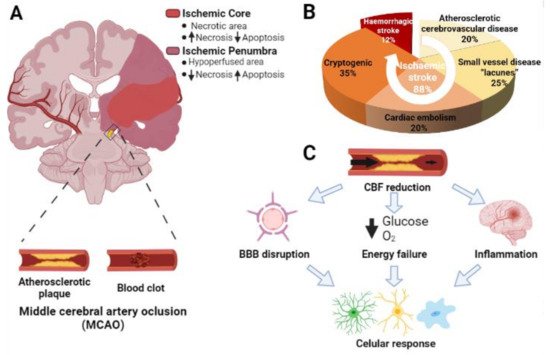
Microglia represent 10–15% of the total cells in the brain. These cells are considered as the resident macrophages and the first defense line in the central nervous system (CNS) against pathogens [15][14]. In physiological conditions, microglia have a high capacity to respond to changes in the CNS microenvironment owing to their processes [16][15]. These changes may be triggered by microorganisms such as bacteria or viruses, but also by neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, or cerebral ischemia. Particularly, microglia activation is one of the first events that occurs after an insult such as brain ischemia [17][16], taking place from minutes to a few hours after the start of the episode [18][17] (Figure 2).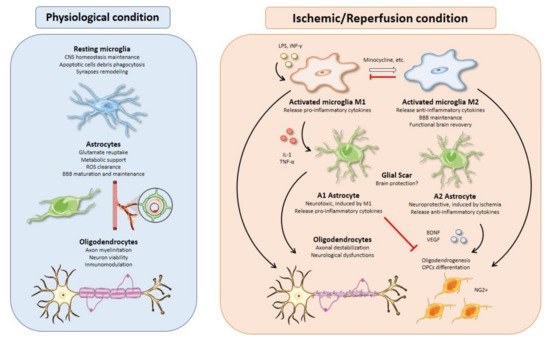
2.1. Microglial Activation Timing upon Brain Ischemia
2.2. M1 versus M2 Microglial Profiles
2.3. Inductors of Microglial Activation in Stroke
2.4. Therapies Based on Activated Microglia
3. Astrocytes
Astrocytes account for 50% of the human brain volume [70][69] and are normally classified into two mayor types according to morphological and spatial criteria: fibrous astrocytes in the white matter and protoplasmic astrocytes predominant in the grey matter [71][70]. The functional implications of each cell type in ischemic stroke are beyond the scope of this review, so the global term “astrocyte” will be used herein to encompass this complexity. Astrocytes have long been considered as a mere buffering system to sustain the correct functioning of the neuronal circuitry. However, in light of the evidence accumulated along the last decades, it becomes clear that astrocytes are active players in every physiological functions of the CNS as well as in pathological events following ischemic injury.3.1. Excitotoxicity Modulation
3.2. Astrocyte-Neuron Metabolic Relationships in Stroke
3.3. Oxidative Stress Management
References
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics—2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743.
- Neurology, T.L. A unified European action plan on stroke. Lancet Neurol. 2020, 19, 963.
- Katan, M.; Luft, A. Global Burden of Stroke. Semin. Neurol. 2018, 38, 208–211.
- Venketasubramanian, N.; Yoon, B.W.; Pandian, J.; Navarro, J.C. Stroke Epidemiology in South, East, and South-East Asia: A Review. J. Stroke 2017, 19, 286–294.
- García-Moncó, J.C.; Cabrera-Muras, A.; Collía-Fernández, A.; Erburu-Iriarte, M.; Rodrigo-Armenteros, P.; Oyarzun-Irazu, I.; Martínez-Condor, D.; Bilbao-González, A.; Carmona-Abellán, M.; Caballero-Romero, I.; et al. Neurological reasons for consultation and hospitalization during the COVID-19 pandemic. Neurol. Sci. 2020, 41, 3031–3038.
- Cagnazzo, F.; Piotin, M.; Escalard, S.; Maier, B.; Ribo, M.; Requena, M.; Pop, R.; Hasiu, A.; Gasparotti, R.; Mardighian, D.; et al. European Multicenter Study of ET-COVID-19. Stroke 2021, 52, 31–39.
- Fifi, J.T.; Mocco, J. COVID-19 related stroke in young individuals. Lancet Neurol. 2020, 19, 713–715.
- Yaghi, S.; Bernstein, R.A.; Passman, R.; Okin, P.M.; Furie, K.L. Cryptogenic Stroke. Circ. Res. 2017, 120, 527–540.
- Sarmah, D.; Banerjee, M.; Datta, A.; Kalia, K.; Dhar, S.; Yavagal, D.R.; Bhattacharya, P. Nanotechnology in the diagnosis and treatment of stroke. Drug Discov. Today 2020, 26, 585–592.
- Meschia, J.F.; Brott, T. Ischaemic stroke. Eur. J. Neurol. 2018, 25, 35–40.
- Campbell, B.C.V.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70.
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385.
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397.
- Picard, K.; St-Pierre, M.K.; Vecchiarelli, H.A.; Bordeleau, M.; Tremblay, M. Neuroendocrine, neuroinflammatory and pathological outcomes of chronic stress: A story of microglial remodeling. Neurochem. Int. 2021, 145, 104987.
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318.
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789.
- Nakajima, K.; Kohsaka, S. Microglia: Neuroprotective and neurotrophic cells in the central nervous system. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2004, 4, 65–84.
- Morioka, T.; Kalehua, A.N.; Streit, W.J. Characterization of microglial reaction after middle cerebral artery occlusion in rat brain. J. Comp. Neurol. 1993, 327, 123–132.
- Schroeter, M.; Jander, S.; Huitinga, I.; Witte, O.W.; Stoll, G. Phagocytic response in photochemically induced infarction of rat cerebral cortex. The role of resident microglia. Stroke 1997, 28, 382–386.
- Rupalla, K.; Allegrini, P.R.; Sauer, D.; Wiessner, C. Time course of microglia activation and apoptosis in various brain regions after permanent focal cerebral ischemia in mice. Acta Neuropathol. 1998, 96, 172–178.
- Gorlamandala, N.; Parmar, J.; Craig, A.J.; Power, J.M.; Moorhouse, A.J.; Krishnan, A.V.; Housley, G.D. Focal Ischaemic Infarcts Expand Faster in Cerebellar Cortex than Cerebral Cortex in a Mouse Photothrombotic Stroke Model. Transl. Stroke Res. 2018, 9, 643–653.
- Neher, J.J.; Emmrich, J.V.; Fricker, M.; Mander, P.K.; Théry, C.; Brown, G.C. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc. Natl. Acad. Sci. USA 2013, 110, E4098–E4107.
- Perego, C.; Fumagalli, S.; De Simoni, M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflamm. 2011, 8, 174.
- Jin, X.; Yamashita, T. Microglia in central nervous system repair after injury. J. Biochem. 2016, 159, 491–496.
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758.
- Prinz, M.; Mildner, A. Microglia in the CNS: Immigrants from another world. Glia 2011, 59, 177–187.
- Ma, Y.M.; Li, C.D.; Zhu, Y.B.; Xu, X.; Li, J.; Cao, H. The mechanism of RvD1 alleviates type 2 diabetic neuropathic pain by influencing microglia polarization in rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2017, 33, 277–281.
- Morrison, H.W.; Filosa, J.A. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflamm. 2013, 10, 4.
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13.
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell Physiol. 2018, 233, 6425–6440.
- Colton, C.A. Heterogeneity of microglial activation in the innate immune response in the brain. J. Neuroimmune Pharmacol. 2009, 4, 399–418.
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783.
- Lyu, J.; Xie, D.; Bhatia, T.N.; Leak, R.K.; Hu, X.; Jiang, X. Microglial/Macrophage polarization and function in brain injury and repair after stroke. CNS Neurosci. Ther. 2021, 27, 515–527.
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135.
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42.
- Qin, C.; Zhou, L.Q.; Ma, X.T.; Hu, Z.W.; Yang, S.; Chen, M.; Bosco, D.B.; Wu, L.J.; Tian, D.S. Dual Functions of Microglia in Ischemic Stroke. Neurosci. Bull. 2019, 35, 921–933.
- Gaojian, T.; Dingfei, Q.; Linwei, L.; Xiaowei, W.; Zheng, Z.; Wei, L.; Tong, Z.; Benxiang, N.; Yanning, Q.; Wei, Z.; et al. Parthenolide promotes the repair of spinal cord injury by modulating M1/M2 polarization via the NF-κB and STAT 1/3 signaling pathway. Cell Death Discov. 2020, 6, 97.
- Yang, F.; Li, W.B.; Qu, Y.W.; Gao, J.X.; Tang, Y.S.; Wang, D.J.; Pan, Y.J. Bone marrow mesenchymal stem cells induce M2 microglia polarization through PDGF-AA/MANF signaling. World J. Stem Cells 2020, 12, 633–658.
- Parisi, C.; Napoli, G.; Pelegrin, P.; Volonté, C. M1 and M2 Functional Imprinting of Primary Microglia: Role of P2X7 Activation and miR-125b. Mediat. Inflamm. 2016, 2016, 2989548.
- Tu, H.; Chu, H.; Guan, S.; Hao, F.; Xu, N.; Zhao, Z.; Liang, Y. The role of the M1/M2 microglia in the process from cancer pain to morphine tolerance. Tissue Cell 2021, 68, 101438.
- Lana, D.; Ugolini, F.; Nosi, D.; Wenk, G.L.; Giovannini, M.G. The Emerging Role of the Interplay Among Astrocytes, Microglia, and Neurons in the Hippocampus in Health and Disease. Front. Aging Neurosci. 2021, 13, 651973.
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194.
- Dabrowska, S.; Andrzejewska, A.; Lukomska, B.; Janowski, M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J. Neuroinflamm. 2019, 16, 178.
- Gilchrist, S.E.; Goudarzi, S.; Hafizi, S. Gas6 Inhibits Toll-Like Receptor-Mediated Inflammatory Pathways in Mouse Microglia via Axl and Mer. Front. Cell Neurosci. 2020, 14, 576650.
- Lu, Y.; Li, X.; Liu, S.; Zhang, Y.; Zhang, D. Toll-like Receptors and Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 72.
- Pan, W.; Kastin, A.J. Tumor necrosis factor and stroke: Role of the blood-brain barrier. Prog. Neurobiol. 2007, 83, 363–374.
- Welser, J.V.; Li, L.; Milner, R. Microglial activation state exerts a biphasic influence on brain endothelial cell proliferation by regulating the balance of TNF and TGF-beta1. J. Neuroinflamm. 2010, 7, 89.
- Carvalho, T.G.; Alves-Silva, J.; de Souza, J.M.; Real, A.; Doria, J.G.; Vieira, E.L.M.; Gomes, G.F.; de Oliveira, A.C.; Miranda, A.S.; Ribeiro, F.M. Metabotropic glutamate receptor 5 ablation accelerates age-related neurodegeneration and neuroinflammation. Neurochem. Int. 2019, 126, 218–228.
- Rahman, M.S.; Yang, J.; Luan, Y.; Qiu, Z.; Zhang, J.; Lu, H.; Chen, X.; Liu, Y. Attenuation of Acute Intracerebral Hemorrhage-Induced Microglial Activation and Neuronal Death Mediated by the Blockade of Metabotropic Glutamate Receptor 5 In Vivo. Neurochem. Res. 2020, 45, 1230–1243.
- Kaiser, M.; Penk, A.; Franke, H.; Krügel, U.; Nörenberg, W.; Huster, D.; Schaefer, M. Lack of functional P2X7 receptor aggravates brain edema development after middle cerebral artery occlusion. Purinergic Signal. 2016, 12, 453–463.
- Nguyen, H.M.; di Lucente, J.; Chen, Y.J.; Cui, Y.; Ibrahim, R.H.; Pennington, M.W.; Jin, L.W.; Maezawa, I.; Wulff, H. Biophysical basis for Kv1.3 regulation of membrane potential changes induced by P2X4-mediated calcium entry in microglia. Glia 2020, 68, 2377–2394.
- Hou, K.; Li, G.; Yu, J.; Xu, K.; Wu, W. Receptors, Channel Proteins, and Enzymes Involved in Microglia-mediated Neuroinflammation and Treatments by Targeting Microglia in Ischemic Stroke. Neuroscience 2021, 460, 167–180.
- Chen, Y.; Yu, S.; Wu, Q.; Tang, Y.; Jiang, C.; Zhuang, Z.; Zhao, N.; Huang, B.; Xie, L. Effect of bone marrow mesenchymal stem cells conditioned medium on microglia and its secretion of arginase 1 in rats. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 2017, 31, 1500–1505.
- Lu, Y.; Zhou, M.; Li, Y.; Li, Y.; Hua, Y.; Fan, Y. Minocycline promotes functional recovery in ischemic stroke by modulating microglia polarization through STAT1/STAT6 pathways. Biochem. Pharmacol. 2021, 186, 114464.
- Serhan, A.; Aerts, J.L.; Boddeke, E.; Kooijman, R. Neuroprotection by Insulin-like Growth Factor-1 in Rats with Ischemic Stroke is Associated with Microglial Changes and a Reduction in Neuroinflammation. Neuroscience 2020, 426, 101–114.
- Scholz, R.; Sobotka, M.; Caramoy, A.; Stempfl, T.; Moehle, C.; Langmann, T. Minocycline counter-regulates pro-inflammatory microglia responses in the retina and protects from degeneration. J. Neuroinflamm. 2015, 12, 209.
- Tomás-Camardiel, M.; Rite, I.; Herrera, A.J.; de Pablos, R.M.; Cano, J.; Machado, A.; Venero, J.L. Minocycline reduces the lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption of the blood-brain barrier, and damage in the nigral dopaminergic system. Neurobiol. Dis. 2004, 16, 190–201.
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015, 129, 279–295.
- Lampl, Y.; Boaz, M.; Gilad, R.; Lorberboym, M.; Dabby, R.; Rapoport, A.; Anca-Hershkowitz, M.; Sadeh, M. Minocycline treatment in acute stroke: An open-label, evaluator-blinded study. Neurology 2007, 69, 1404–1410.
- Kikuchi, K.; Tanaka, E.; Murai, Y.; Tancharoen, S. Clinical trials in acute ischemic stroke. CNS Drugs 2014, 28, 929–938.
- Liu, Z.J.; Ran, Y.Y.; Qie, S.Y.; Gong, W.J.; Gao, F.H.; Ding, Z.T.; Xi, J.N. Melatonin protects against ischemic stroke by modulating microglia/macrophage polarization toward anti-inflammatory phenotype through STAT3 pathway. CNS Neurosci. Ther. 2019, 25, 1353–1362.
- Zhou, J.M.; Gu, S.S.; Mei, W.H.; Zhou, J.; Wang, Z.Z.; Xiao, W. Ginkgolides and bilobalide protect BV2 microglia cells against OGD/reoxygenation injury by inhibiting TLR2/4 signaling pathways. Cell Stress Chaperones 2016, 21, 1037–1053.
- Xi, Z.; Xu, C.; Chen, X.; Wang, B.; Zhong, Z.; Sun, Q.; Sun, Y.; Bian, L. Protocatechuic Acid Suppresses Microglia Activation and Facilitates M1 to M2 Phenotype Switching in Intracerebral Hemorrhage Mice. J. Stroke Cereb. Dis. 2021, 30, 105765.
- Li, D.; Wang, C.; Yao, Y.; Chen, L.; Liu, G.; Zhang, R.; Liu, Q.; Shi, F.D.; Hao, J. mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. FASEB J. 2016, 30, 3388–3399.
- Zhou, X.; Lu, W.; Wang, Y.; Li, J.; Luo, Y. A20-Binding Inhibitor of NF-κB 1 Ameliorates Neuroinflammation and Mediates Antineuroinflammatory Effect of Electroacupuncture in Cerebral Ischemia/Reperfusion Rats. Evid. Based Complement. Altern. Med. 2020, 2020, 6980398.
- Li, F.; Ma, Q.; Zhao, H.; Wang, R.; Tao, Z.; Fan, Z.; Zhang, S.; Li, G.; Luo, Y. L-3-n-Butylphthalide reduces ischemic stroke injury and increases M2 microglial polarization. Metab. Brain Dis. 2018, 33, 1995–2003.
- Liu, W.; Wang, X.; Yang, S.; Huang, J.; Xue, X.; Zheng, Y.; Shang, G.; Tao, J.; Chen, L. Electroacupunctre improves motor impairment via inhibition of microglia-mediated neuroinflammation in the sensorimotor cortex after ischemic stroke. Life Sci. 2016, 151, 313–322.
- Ma, Z.; Zhang, Z.; Bai, F.; Jiang, T.; Yan, C.; Wang, Q. Electroacupuncture Pretreatment Alleviates Cerebral Ischemic Injury Through α7 Nicotinic Acetylcholine Receptor-Mediated Phenotypic Conversion of Microglia. Front. Cell Neurosci. 2019, 13, 537.
- Mason, S. Lactate Shuttles in Neuroenergetics-Homeostasis, Allostasis and Beyond. Front. Neurosci. 2017, 11, 43.
- Liu, Z.; Chopp, M. Astrocytes, therapeutic targets for neuroprotection and neurorestoration in ischemic stroke. Prog. Neurobiol. 2016, 144, 103–120.
- Chisholm, N.C.; Henderson, M.L.; Selvamani, A.; Park, M.J.; Dindot, S.; Miranda, R.C.; Sohrabji, F. Histone methylation patterns in astrocytes are influenced by age following ischemia. Epigenetics 2015, 10, 142–152.
- Yamada, T.; Kawahara, K.; Kosugi, T.; Tanaka, M. Nitric oxide produced during sublethal ischemia is crucial for the preconditioning-induced down-regulation of glutamate transporter GLT-1 in neuron/astrocyte co-cultures. Neurochem. Res. 2006, 31, 49–56.
- Zeevalk, G.D.; Davis, N.; Hyndman, A.G.; Nicklas, W.J. Origins of the extracellular glutamate released during total metabolic blockade in the immature retina. J. Neurochem. 1998, 71, 2373–2381.
- Ouyang, Y.B.; Voloboueva, L.A.; Xu, L.J.; Giffard, R.G. Selective dysfunction of hippocampal CA1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. J. Neurosci. 2007, 27, 4253–4260.
- Harvey, B.K.; Airavaara, M.; Hinzman, J.; Wires, E.M.; Chiocco, M.J.; Howard, D.B.; Shen, H.; Gerhardt, G.; Hoffer, B.J.; Wang, Y. Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS ONE 2011, 6, e22135.
- Weller, M.L.; Stone, I.M.; Goss, A.; Rau, T.; Rova, C.; Poulsen, D.J. Selective overexpression of excitatory amino acid transporter 2 (EAAT2) in astrocytes enhances neuroprotection from moderate but not severe hypoxia-ischemia. Neuroscience 2008, 155, 1204–1211.
- Shen, Y.; He, P.; Fan, Y.Y.; Zhang, J.X.; Yan, H.J.; Hu, W.W.; Ohtsu, H.; Chen, Z. Carnosine protects against permanent cerebral ischemia in histidine decarboxylase knockout mice by reducing glutamate excitotoxicity. Free Radic. Biol. Med. 2010, 48, 727–735.
- Cui, X.; Li, L.; Hu, Y.Y.; Ren, S.; Zhang, M.; Li, W.B. Sulbactam Plays Neuronal Protective Effect Against Brain Ischemia via Upregulating GLT1 in Rats. Mol. Neurobiol. 2015, 51, 1322–1333.
- Rao, V.L.; Dogan, A.; Todd, K.G.; Bowen, K.K.; Kim, B.T.; Rothstein, J.D.; Dempsey, R.J. Antisense knockdown of the glial glutamate transporter GLT-1, but not the neuronal glutamate transporter EAAC1, exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J. Neurosci. 2001, 21, 1876–1883.
- Yang, J.; Vitery, M.D.C.; Chen, J.; Osei-Owusu, J.; Chu, J.; Qiu, Z. Glutamate-Releasing SWELL1 Channel in Astrocytes Modulates Synaptic Transmission and Promotes Brain Damage in Stroke. Neuron 2019, 102, 813–827.e6.
- Zhang, Y.; Jin, Y.; Behr, M.J.; Feustel, P.J.; Morrison, J.P.; Kimelberg, H.K. Behavioral and histological neuroprotection by tamoxifen after reversible focal cerebral ischemia. Exp. Neurol. 2005, 196, 41–46.
- Duan, S.; Anderson, C.M.; Keung, E.C.; Chen, Y.; Chen, Y.; Swanson, R.A. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 2003, 23, 1320–1328.
- Liang, Z.; Wang, X.; Hao, Y.; Qiu, L.; Lou, Y.; Zhang, Y.; Ma, D.; Feng, J. The Multifaceted Role of Astrocyte Connexin 43 in Ischemic Stroke Through Forming Hemichannels and Gap Junctions. Front. Neurol. 2020, 11, 703.
- Deng, Z.H.; Liao, J.; Zhang, J.Y.; Liang, C.; Song, C.H.; Han, M.; Wang, L.H.; Xue, H.; Zhang, K.; Zabeau, L.; et al. Inhibition of the connexin 43 elevation may be involved in the neuroprotective activity of leptin against brain ischemic injury. Cell Mol. Neurobiol. 2014, 34, 871–879.
- Turner, D.A.; Adamson, D.C. Neuronal-astrocyte metabolic interactions: Understanding the transition into abnormal astrocytoma metabolism. J. Neuropathol. Exp. Neurol. 2011, 70, 167–176.
- MacVicar, B.A.; Newman, E.A. Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect. Biol. 2015, 7.
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629.
- Dienel, G.A. Lack of appropriate stoichiometry: Strong evidence against an energetically important astrocyte-neuron lactate shuttle in brain. J. Neurosci. Res. 2017, 95, 2103–2125.
- Hertz, L.; Chen, Y. Glycogenolysis, an Astrocyte-Specific Reaction, is Essential for Both Astrocytic and Neuronal Activities Involved in Learning. Neuroscience 2018, 370, 27–36.
- Takahashi, S.; Iizumi, T.; Mashima, K.; Abe, T.; Suzuki, N. Roles and regulation of ketogenesis in cultured astroglia and neurons under hypoxia and hypoglycemia. ASN Neuro 2014, 6.
- Guzman, M.; Blazquez, C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol. Metab. 2001, 12, 169–173.
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538.
- Bazzigaluppi, P.; Lake, E.M.; Beckett, T.L.; Koletar, M.M.; Weisspapir, I.; Heinen, S.; Mester, J.; Lai, A.; Janik, R.; Dorr, A.; et al. Imaging the Effects of beta-Hydroxybutyrate on Peri-Infarct Neurovascular Function and Metabolism. Stroke 2018, 49, 2173–2181.
- Gibson, C.L.; Murphy, A.N.; Murphy, S.P. Stroke outcome in the ketogenic state--a systematic review of the animal data. J. Neurochem. 2012, 123 (Suppl. 2), 52–57.
- Yin, J.; Han, P.; Tang, Z.; Liu, Q.; Shi, J. Sirtuin 3 mediates neuroprotection of ketones against ischemic stroke. J. Cereb. Blood Flow Metab. 2015, 35, 1783–1789.
- Zhou, L.; Deepa, S.S.; Etzler, J.C.; Ryu, J.; Mao, X.; Fang, Q.; Liu, D.D.; Torres, J.M.; Jia, W.; Lechleiter, J.D.; et al. Adiponectin activates AMP-activated protein kinase in muscle cells via APPL1/LKB1-dependent and phospholipase C/Ca2+/Ca2+/calmodulin-dependent protein kinase kinase-dependent pathways. J. Biol. Chem. 2009, 284, 22426–22435.
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503.
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781.
- Jimenez-Blasco, D.; Santofimia-Castano, P.; Gonzalez, A.; Almeida, A.; Bolanos, J.P. Astrocyte NMDA receptors’ activity sustains neuronal survival through a Cdk5-Nrf2 pathway. Cell Death Differ. 2015, 22, 1877–1889.
- Takagi, T.; Kitashoji, A.; Iwawaki, T.; Tsuruma, K.; Shimazawa, M.; Yoshimura, S.; Iwama, T.; Hara, H. Temporal activation of Nrf2 in the penumbra and Nrf2 activator-mediated neuroprotection in ischemia-reperfusion injury. Free Radic. Biol. Med. 2014, 72, 124–133.
- Skowronska, K.; Obara-Michlewska, M.; Zielinska, M.; Albrecht, J. NMDA Receptors in Astrocytes: In Search for Roles in Neurotransmission and Astrocytic Homeostasis. Int. J. Mol. Sci. 2019, 20, 309.
- Dzamba, D.; Honsa, P.; Valny, M.; Kriska, J.; Valihrach, L.; Novosadova, V.; Kubista, M.; Anderova, M. Quantitative Analysis of Glutamate Receptors in Glial Cells from the Cortex of GFAP/EGFP Mice Following Ischemic Injury: Focus on NMDA Receptors. Cell Mol. Neurobiol. 2015, 35, 1187–1202.
- Wang, H.; Yan, H.; Zhang, S.; Wei, X.; Zheng, J.; Li, J. The GluN3A subunit exerts a neuroprotective effect in brain ischemia and the hypoxia process. ASN Neuro 2013, 5, 231–242.
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215.
- Juliana, C.; Fernandes-Alnemri, T.; Kang, S.; Farias, A.; Qin, F.; Alnemri, E.S. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J. Biol. Chem. 2012, 287, 36617–36622.
- Abais, J.M.; Xia, M.; Zhang, Y.; Boini, K.M.; Li, P.L. Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal. 2015, 22, 1111–1129.
- Xu, N.; Zhang, Y.; Doycheva, D.M.; Ding, Y.; Zhang, Y.; Tang, J.; Guo, H.; Zhang, J.H. Adiponectin attenuates neuronal apoptosis induced by hypoxia-ischemia via the activation of AdipoR1/APPL1/LKB1/AMPK pathway in neonatal rats. Neuropharmacology 2018, 133, 415–428.
- Duan, J.; Yin, Y.; Cui, J.; Yan, J.; Zhu, Y.; Guan, Y.; Wei, G.; Weng, Y.; Wu, X.; Guo, C.; et al. Chikusetsu Saponin IVa Ameliorates Cerebral Ischemia Reperfusion Injury in Diabetic Mice via Adiponectin-Mediated AMPK/GSK-3beta Pathway In Vivo and In Vitro. Mol. Neurobiol. 2016, 53, 728–743.
- Liu, H.; Wu, X.; Luo, J.; Zhao, L.; Li, X.; Guo, H.; Bai, H.; Cui, W.; Guo, W.; Feng, D.; et al. Adiponectin peptide alleviates oxidative stress and NLRP3 inflammasome activation after cerebral ischemia-reperfusion injury by regulating AMPK/GSK-3beta. Exp. Neurol. 2020, 329, 113302.
- Allaman, I.; Belanger, M.; Magistretti, P.J. Astrocyte-neuron metabolic relationships: For better and for worse. Trends Neurosci. 2011, 34, 76–87.
- Sarkar, S.; Mukherjee, A.; Swarnakar, S.; Das, N. Nanocapsulated Ascorbic Acid in Combating Cerebral Ischemia Reperfusion- Induced Oxidative Injury in Rat Brain. Curr. Alzheimer Res. 2016, 13, 1363–1373.
- Peng, L.; Zhao, Y.; Li, Y.; Zhou, Y.; Li, L.; Lei, S.; Yu, S.; Zhao, Y. Effect of DJ-1 on the neuroprotection of astrocytes subjected to cerebral ischemia/reperfusion injury. J. Mol. Med. 2019, 97, 189–199.
- Guo, X.; Jiang, Q.; Tuccitto, A.; Chan, D.; Alqawlaq, S.; Won, G.J.; Sivak, J.M. The AMPK-PGC-1alpha signaling axis regulates the astrocyte glutathione system to protect against oxidative and metabolic injury. Neurobiol. Dis. 2018, 113, 59–69.
- Trendelenburg, G.; Prass, K.; Priller, J.; Kapinya, K.; Polley, A.; Muselmann, C.; Ruscher, K.; Kannbley, U.; Schmitt, A.O.; Castell, S.; et al. Serial analysis of gene expression identifies metallothionein-II as major neuroprotective gene in mouse focal cerebral ischemia. J. Neurosci. 2002, 22, 5879–5888.