Immunophilins are a family of proteins whose signature domain is the peptidylprolyl-isomerase domain. High molecular weight immunophilins are characterized by the additional presence of tetratricopeptide-repeats (TPR) through which they bind to the 90-kDa heat-shock protein (Hsp90), and via this chaperone, immunophilins contribute to the regulation of the biological functions of several client-proteins. Among these Hsp90-binding immunophilins, there are two highly homologous members named FKBP51 and FKBP52 (FK506-binding protein of 51-kDa and 52-kDa, respectively) that were first characterized as components of the Hsp90-based heterocomplex associated to steroid receptors. Afterwards, they emerged as likely contributors to a variety of other hormone-dependent diseases, stress-related pathologies, psychiatric disorders, cancer, and other syndromes characterized by misfolded proteins. The differential biological actions of these immunophilins have been assigned to the structurally similar, but functionally divergent enzymatic domain.
- FKBP51
- FKBP52
- Hsp90
- dynein
- telomerase
- NF-κB
- neurodifferentiation
- cell differentiation
1. Introduction
2. General Aspects of FKPB51 and FKBP52
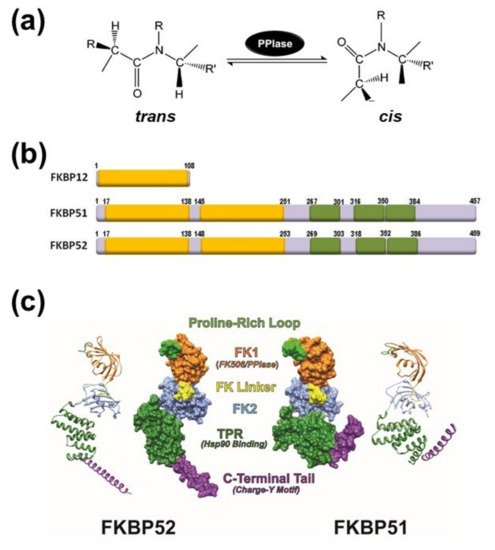
The immunosuppressive action of FK506 or cyclosporine A is responsibility of the drug binding to the smallest members of both classical immunophilin subfamilies, i.e. FKBP12, encoded by the FKBP1A gene, and cyclophilin A (CyPA), encoded by the PP1A gene. As consequence of these specific interactions, the liganded immunophilin (but not the drug or the protein alone) impairs the activity of calcineurin [7][13], a Ser/Thr-phosphatase also known as PP2B. Thus, the transcription factor NFAT (Nuclear Factor of Activated T-cells) remains phosphorylated in the cytoplasm of lymphocytes, and the production of interleukines and interferon-γ is consequently avoided (see [8][14] for a comprehensive review for this mechanism). On the other hand, members of the immunophilin family that possess higher molecular weight show more complex protein architecture because they have additional domains to the PPIase domain. One of the best characterized immunophilins is the 52-kDa FK506-binding protein, FKBP52 (gene name FKBP4)[9][15], which is the archetype of the Hsp90-binding subfamily (Figure 1b). In addition to the PPIase domain (also called FKBD1 or FK1 domain), FKBP52 shows three repetitions in tandem of a degenerative sequence of 34 amino acids named tetratricopeptide repeats (TPR), which has the capability to form associations with Hsp90 dimers [10][16]. The Hsp90-binding immunophilin FKBP52 shows a close-related partner, FKBP51 (gene name FKBP5), with whom it shares 75% similarity and 60% identity. Due to this high homology, but different conformational features (Figure 1c), both immunophilins usually compete one another for binding and functional properties of client-proteins, although this antagonistic effect shows exceptions in a few numbers of cases and their biological effects become redundant (see afterwards). Both immunophilins possess a short sequence of 7 to 9 amino acids that forms an FK-linker region. It connects the FK1 domain with the FK2 region. The linker sequence of FKBP52 is capped by a TEEED phosphorylation sequence that is substrate of casein kinase-2 (CK2). The resultant phosphorylation at Thr143 impairs the FKBP52•Hsp90 interaction [11][17] and also abrogates the normal regulation seen on steroid receptors by FKBP52. In the case of FKBP51, this loop is capped by FED, a conserved sequence in high molecular weight immunophilins, such that the phosphorylation of the site by CK2 does not occur. Nevertheless, these differences cannot account for the lack of receptor potentiation capability shown by FKBP51, which is most of the times an inhibitor of the biological actions of nuclear receptors. As it was stated above, it is accepted that the FK1 domains of both immunophilins are the major structural elements responsible for the divergent properties of FKBP51 and FKBP52 on the steroid receptor action [12][13][14][15][18,19,20,21]. Nevertheless, while the PPIase domain is important in this regard, it appears that the enzymatic activity of prolyl-isomerase is not always essential [14][16][20,22]. The immunophilin is often part of a heterocomplex with Hsp90, such that the interaction with the chaperone also influences the FK1 domain of FKBPs [17][23]. The binding of Hsp90 to the immunophilin enables the FK1 domain to interact with the ligand binding domain of the glucocorticoid receptor (GR), thereby influencing the GR conformation and the steroid binding affinity [18][24]. Inasmuch as the conformations of the FK1 domains of both FKBPs differ around the PPIase pocket, it is not surprising that this fact causes differential protein interactions with client-factors [19][20][25,26]. The structure of the FK2 region (second yellow box in Figure 1b) is similar to that of the FK1 region, but FK2 does not show enzymatic activity of isomerase and cannot recognize immunosuppressive ligands. When the FK2 region of FKBP51 was modified by point mutations, Hsp90 binding took place yet the mutant cannot integrate normally into the receptor heterocomplex [19][25]. Therefore, it might be possible that the mutation impairs key interactions not only with other members of the receptor heterocomplex, but also with the receptor itself. In contrast to the FK1 domain of FKBP52, the FK1 domain of FKBP51 does not show stimulating activity on steroid receptor activity, but random mutagenesis studies evidenced that two key point mutations in the FK1 domain of FKBP51 confer full receptor potentiation activity to this immunophilin equivalent to that observed for FKBP52 [21][27]. This suggestive finding is in line with the notion that both proteins may have diverged during evolution by only a very limited number of modified amino acid residues. Inasmuch as these two residues are in the proline-rich loop (see Figure 1c), it may be inferred that this region of FKBP52 is functionally relevant for the regulation of steroid receptor activity. Accordingly, it has recently been postulated that the loop serves as an interaction surface with the ligand binding domain of the receptor [22][23][28,29]. The TPR domain located at the C-terminal end of the immunophilin confers the capability to interact with the chaperone Hsp90 via the C-terminal sequence of the chaperone, the EEVD motif [10][24][16,30]. On the other hand, isothermal titration calorimetry studies performed with FKBP51 and FKBP52 demonstrated that the former immunophilin interacts with Hsp90 dimers with lower affinity (about one third) compared to FKBP52 [25][31]. Nonetheless, due to the influence of the relative abundance of each FKBPs in each cell type, solely the relative affinity of each immunophilin for Hsp90 cannot permit accurately predict the stoichiometry of the oligomeric complexes formed with the receptors.3. Immunophilins Play a Key Role in Protein Trafficking
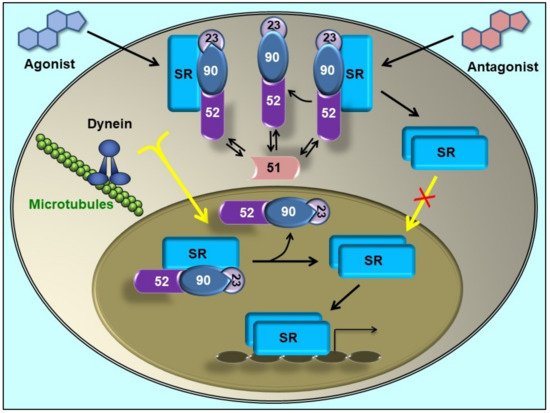
Steroid receptors bind their cognate ligands only if they are assembled with Hsp90 in oligomeric structures [26][27][32,33], this Hsp90-based complex being a biological on/off functional switch. This property agrees with the properties of Hsp90, a chaperone that at variance of others that prefer partially unfolded clients, favours substrates that possess a preserved tertiary structure. This is particularly notorious for some members of the steroid receptor family such as GR, mineralocorticoid receptor (MR), progesterone receptor (PR) and androgen receptor (AR) [28][34]. They are transcription factors with properties of phosphoproteins [29][30][31][35,36,37], and are activated by ligand binding. Steroid receptors are assembled with molecular chaperones and co-chaperones, including Hsp90, Hsp70, Hsp40, p23 and a TPR-domain protein, usually a high molecular weight immunophilin such as FKBP51, FKBP52, CyP40 or PP5. The interaction of TPR proteins with Hsp90 (and Hsp70) is conserved in nature and broadly distributed in both animal and plant kingdoms [32][33][34][38,39,40]. The discovery that the dynein/dynactin motor complex co-immunoprecipitates with GR [35][41] and MR [36][42] via its association to the PPIase domain of FKBP52 modified the classic view for the mechanism of steroid receptor activation. In the absence of steroid, some receptors such as GR, MR or AR (which also interacts with dynein/dynactin, as it was further demonstrated [37][43]) are primarily cytoplasmic proteins. Upon steroid binding, they rapidly accumulate in the nucleus. Other receptors such as the estrogen receptor (ER) are constitutively nuclear even in the absence of steroid, but they are not statically confined to a cell compartment, but are continuously shuttling between cytoplasm and nucleus [38][39][40][41][44,45,46,47]. Classically, the driving force for soluble protein movement throughout the cytoplasm was always assumed to occur by simple diffusion, and steroid receptors were not the exception. The classic model for receptor activation posited in the ‘80s proposed that Hsp90 should dissociate from the receptor upon steroid binding to release the transcription factor from the cytoplasmic anchorage sites. When the key role for FKBP52 in the receptor retrograde movement was demonstrated, the classic dogma was replaced by a model where the entire receptor•Hsp90•FKBP52•dynein complex moves throughout the cytoplasm, translocates intact through the nuclear pore, and is finally dissociated in the nucleoplasm [36][42][42,48] (Figure 2). A direct corollary of this novel model is the prediction that receptor dimerization cannot be a cytoplasmic event because the association of the receptor with the Hsp90 chaperone complex (a key requirement for receptor retrotransport) blocks the dimerization domain; therefore, it may be predicted that the oligomeric chaperone complex should be released in the nucleoplasm after the translocation through the nuclear pore, which would allow receptor dimerization. This prediction was effectively demonstrated time after for the GR [43][49] and the MR [44][50] using different methodologies.