The molecular characterization of CTCs could provide the information on phenotypic identification of malignant cells and genetic alteration that may change according to disease progression and therapy resistance (as illustrated in). However, monitoring CTC characteristics was initially performed on enriched fractions, which provided only very limited information on tumor heterogeneity. With the advances in technologies for single-cell analysis made during the past 5 years, analyses of CTCs at single-cell resolution in peripheral blood could offer a unique minimally invasive approach to characterize and monitor dynamic changes in tumor heterogeneity in individual patients with cancer at the genomic, transcriptomic, proteomic, and functional levels.
- breast cancer metastasis
- genomic analysis
- diagnosis
- prognosis
- therapy
1. Introduction
Breast cancer (BC) is the most common tumor worldwide with around 2,260,000 new diagnoses worldwide in 2020, accounting for approximately 11.7% of all cancers [1]. The 5-year overall survival rate of BC patients without metastasis is 90% [2]; however, distant metastasis can lead to a dramatic reduction of this rate to approximately 25% [3][4]. Great advances in mouse models and genomic sequencing technologies brought new ways into investigating the molecular mechanisms of metastasis. Here, we summarize current findings of molecular mechanisms of BC site-specific metastasis discovered with the advent and power of genomic technologies and discuss about the potential prognostic and therapeutic implications from the current perspective.
2. Metastasis Prediction from Genomic Profiling of Circulating Tumor Cells
Circulating tumor cells (CTCs) are the tumor cells that are released from the formation and growth of primary tumor and/or metastatic sites into the bloodstream. Nevertheless, the environment in the bloodstream is harsh for tumor cells owing to the physical forces, the presence of immune cells, and anoikis, and it is likely that CTCs might undergo a strong selection process which contributes only an extremely small proportion of the CTCs to forming secondary tumors [4][5][6][7][80,81,82,83]. As the dissemination of tumor cells to distant organ sites necessitates a series of processes through the vasculature, it is fostered by close association with activated platelets and macrophages [8][9][10][85,86,87]. The platelet clot then triggers the localization of CD11b+ monocytes/macrophages to the tumor cells, which results in the establishment of microclots (also called CTC clusters) that protect CTCs to survive in blood [11][88].
It is a tool that can be used for positive selection of CTCs expressing cytokeratins (cytoskeletal proteins present in epithelial cells) and Patients with ≥5 CTCs per 7.5 mL of whole blood, as compared with that of the group with <5 CTCs, had a shorter median progression-free survival (2.7 months vs. 7.0 months,p< 0.001) and shorter overall survival (10.1 months vs. >18 months,p< 0.001). The number of CTCs is an independent predictor of progression-free survival and overall survival in patients with MBC [12][97]. Moreover, recent data confirmed the prognostic value of CTCs in early-stage BC [13][14][99,100].
The molecular characterization of CTCs could provide the information on phenotypic identification of malignant cells and genetic alteration that may change according to disease progression and therapy resistance (as illustrated in Figure 12). However, monitoring CTC characteristics was initially performed on enriched fractions [15][103], which provided only very limited information on tumor heterogeneity. With the advances in technologies for single-cell analysis made during the past 5 years, analyses of CTCs at single-cell resolution in peripheral blood could offer a unique minimally invasive approach to characterize and monitor dynamic changes in tumor heterogeneity in individual patients with cancer at the genomic, transcriptomic, proteomic, and functional levels [16][104].
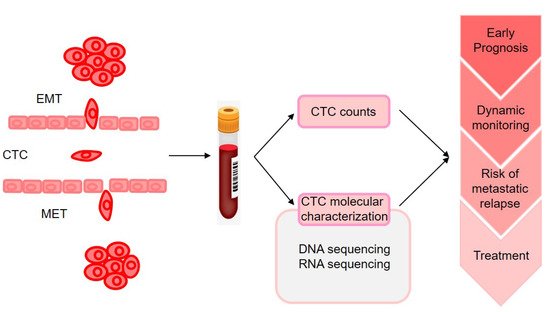
CTCs can be detected at a low cutoff of 1 cell in 27% of patients at the early stage of BC [13][17][99,105], and CTC detection before and/or after neoadjuvant chemotherapy was significantly associated with early metastatic relapse. Hence, the detection and molecular characterization of CTCs in early-stage BC may contribute to the decision-making of clinicians in the selection of patients for strict follow ups for consideration of secondary adjuvant treatments [18][106]. They found that CTCs persisting even months after tumor resection shared several CNAs with matched tumor tissue, suggesting the presence of regions potentially associated with their persistence. In another study, CTCs and primary tumors were profiled using RNA-seq.
Not every CTC will arrive at a secondary site, and even fewer will be able to develop into metastases. There must be different populations of CTCs, some more adept at survival than others, and some with a greater propensity to metastasize than others. How-ever, certain homogeneous genomic gains were detected at primary breast carcinomas and CTCs in MBC, highlighting occult target changes that could be responsible for the preferential passage of tumor cells into circulation [19][111]. Furthermore, higher number of CTCs was associated with genomic alterations in ESR1, GATA3, CDH1, and CCND1, while lower number of CTCs was associated with CDKN2A alterations in MBC [20][112].
A recent study compared gene expression of sequentially generated CTC-derived xenograft (CDX)-derived cell populations, together with online gene expression arrays, and TCGA databases to discover a CTC-driven, liver metastasis-associated TNBC signature [21][114]. This investigation predicted 16 hub genes, 6 biomarkers with clinically available targeted drugs, and 22 survival genes. There is very limited data on the BC organ-specific metastasis based on detection of CTCs [22][23][115,116]. Future mechanistic investigations and prospective studies are needed to delineate the role of CTCs detection in BC organ-specific metastasis.
3. Functional Genomic Analysis of Site-Specific Metastasis of BC
Although sequencing of primary BC provided insight into the biology of early malignancy, the majority of the patients presenting with such a disease will not relapse after conventional therapy. Therefore, understanding the biology of early BC will not help in deciphering the specificities of the lethal disease or translate into treatment advances. Many studies revealed that metastases are clonally related to the primary tumor, sharing many of the driver mutations, but nonetheless have typically acquired additional variants not detectable in the primary lesion during progression [24][25][26][27][28][29][30][31][32][33][34][27,29,71,117,118,119,120,121,122,123,124]. For example, ESR1 mutations or amplification are rarely observed in primary disease but could be acquired during the disease evolution and are prominent and critical drivers of resistance to endocrine therapy [35][36][37][125,126,127].
A further complexity is that metastatic tumor deposits are not exact replicas of the primary tumor from which they arose in either a morphological or a molecular sense. Indeed, metastatic tumors at different sites within an individual may display widely disparate features [38][39][40][128,129,130]. Although the accruing sequencing data suggest that all metastases within a patient shared a common ancestor, significant intermetastasis heterogeneity is invariably observed within patients [30][39][41][42][43][120,129,131,132,133]. Here, we describe the advances in understanding of molecular alterations of different tropisms in BC metastasis, including, bone, lung, liver, and brain, respectively (as illustrated in Table 1).
| The Site of Metastasis | Study | Genes | Expression Status |
|---|---|---|---|
| Bone | Latent bone metastasis in breast cancer tied to Src-dependent survival signals [134] | CXCL12/SDF1; BMP2; IGF2; CXCL14; GMFG; IGF1; JAG1; NOV; PDGFA; PGF; VEGFC; TNFSF10; TGFB1; TGFB3; SPP1; PXDN; CLEC11A; | Upregulated |
| Lung | Genes that mediate breast cancer metastasis to lung [73] | SPARC; IL13RA2; VCAM1; MMP2; MMP1; CXCL1; ID1; COX2; EREG | Upregulated |
| Myeloid progenitor cells in the premetastatic lung promote metastases by inducing mesenchymal to epithelial transition [135] | Versican | Upregulated | |
| NF-κB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression [136] | NF-κB | Downregulated | |
| Liver | Prognosis and Genomic Landscape of Liver Metastasis in Patients With Breast Cancer [137] | ESR1; AKT1; ERBB2; FGFR4 | Upregulated |
| Transcriptional Profiling of Breast Cancer Metastases Identifies Liver Metastasis-Selective Genes Associated with Adverse Outcome in Luminal A Primary Breast Cancer [138] | MFAP5; CDH11; MMP13; FBN1; MXRA5; SFRP4; COL1A2; DPYSL3; EMP1; COL11A1; SPON1; FNDC1; RUNX2; COL3A1 | Downregulated | |
| Brain | Genes that mediate breast cancer metastasis to the brain [72] | ANGPTL4; PLOD2; COL13A1; COX2; PELI1; MMP1; B4GALT6; HBEGF; CSF3; RGC32; LTBP1; FSCN1; LAMA4; ST6GALNAC5 | Upregulated |
| TNFSF10; RARRES3; SCNN1A; SEPP1 | Downregulated | ||
| Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets [122] | CCNE1; EGFR; MYC; EZH2; PIK3CA | Upregulated | |
| Gene Expression Profiling of Breast Cancer Brain Metastasis [139] | SOX2; OLIG2 | Upregulated | |
| CXCL12; MMP2; MMP11; VCAM1; MME | Downregulated |
Bone metastasis develops in approximately 70% of patients with advanced BC and contributes to significant morbidity due to pain and skeletal related events (SREs) Remodeling occurs constantly in the healthy skeleton to regulate calcium homeostasis to repair damage to the bone and withstand new external stresses to the skeleton. The receptor activator of nuclear factor-kB ligand (RANKL) is a major regulator of bone mass. When BC cells metastasize to the bone, they and their circulating factors may affect bone stromal cell targeting, which disrupts the normal bone homeostasis and starts a vicious cycle.
To identify relevant factors, a study of microarray gene-expression analysis of a cohort of metastasis samples that were surgically removed from BC patients, including 16 metastases from bone, 18 from lung, 19 from brain, and 5 from liver [44][134] identified 17 genes that were expressed in bone metastases at a higher level (>2-fold) than in metastases from other sites. CXCL12 is predominantly produced by various bone stromal cells, and acts as the guardian for tumor cells expressing the receptor, CXCR4 JAG1 is an important mediator of bone metastasis by activating the Notch pathway in bone cells. Notch-ligand jagged in tumor cells promotes survival and proliferation by stimulating IL-6 release from osteoblasts or promoting bone metastasis cytokine TGF-β release during bone destruction [45][142].
Only a small percentage of tumor cells that disseminate via circulation can survive at distant sites and form micrometastases [46][143]. The first barrier before tumor cells entering lung microenvironment is the tight cell-cell junctions caused by continuous endothelial cells. To identify which of the genes in the LMS signature are able to confer growth advantages exclusively in the lung microenvironment, they knocked down various lung metastasis genes that were previously assayed for effects on metastatic behavior. IL13Rα2, SPARC and VCAM1 were found to decrease lung metastatic ability but not orthotopic tumor growth.
After evasion from apoptosis, the mutual regulation between tumor cells and other cells in the lung parenchyma facilitate their proliferation and the formation of de novo niches that support metastatic outgrowth. Gene expression profiling revealed that the myeloid cells from metastatic lungs express versican, an extracellular matrix proteoglycan. Versican stimulated MET of metastatic tumor cells by attenuating phospho-Smad2 levels, which resulted in elevated cell proliferation and accelerated metastases [47][135]. Moreover, Huber et al. found that NF-κB activity was necessary for cells to be maintained in a mesenchymal state, as its inhibition causes reversal of EMT, leading to the formation of lung metastases by H-Ras-transformed epithelial cells [48][136].
In a recent work that analyzed the targeted sequencing results of the Memorial Sloan–Kettering Cancer Center (MSKCC) dataset of liver metastasis and primary BC extracted from the MSK-IMPACT Clinical Sequencing Cohort, mutations specific to the metastasis were enriched within the PI3K-AKT pathway molecules [49][137]. The importance of the activation of the PI3K-AKT pathway in hepatic metastases of colorectal cancer was already demonstrated [50][151]. Moreover, Toy et al. observed that TP53, PIK3CA, and GATA3 also had the most frequent mutations in other metastasis organs of BC, including brain, liver, lung, and bone [51][152]. For example, Zhang et al. reported that in bone metastasis of BC, CXCL12/SDF1 and IGF1 positively selected tumor cell clones with elevated proto-oncogene tyrosine-protein kinase sarcoma (SRC)-activity, which in turn led to PI3K-AKT pathway activation and increased survival [44][134].
Oskarsson et al. surmised that cancer cells that underwent an EMT for metastatic dissemination must traverse the reverse process, a mesenchymal-epithelial transition (MET), to initiate metastatic colonization [52][153]. Other studies also showed that N-cadherin, fibroblast growth factor receptor (FGFR) and MMP-9 boosted proliferation and activated liver metastasis to overcome the suppressive effect of E-cadherin [53][154]. Similarly, the homeobox factor Prrx1 is an EMT inducer confer-ring migratory and invasive properties of BC cells, whereas the subsequent downregulation of Prrx-1 induced an MET that facilitated metastatic colonization without suppressing stem cell traits [54][155]. This observation adds to the picture that EMT/MET-orchestrating processes are essential for metastasis, specifically metastasis of BC to the liver.
Metastatic cells invading the CNS parenchyma must pass the blood-brain barrier (BBB), a semipermeable barrier to metastasis, which is comprised of endothelial cells, astrocytes, and pericytes forming the neurovascular unit [55][156]. per-formed a genome-wide expression analysis of brain metastatic cells and parental cells in a BC brain metastases (BCBM) mouse model, which identified 17 genes that were specifically correlated with brain relapse. ST6GALNAC5 over-expression is able to promote transmigration of BC cells through HUVEC endothelial cells, a brain-like endothelial barrier that mimic the BBB in vitro model. ST6GALNAC5 expression in human cancer cells (MDA-MB-231) resulted in the accumulation of GD1α at the cell surface, leading to a lower degree of adhesion between BC cells in a human BBB model [56][160].
Once infiltrated into the brain parenchyma, cancer cells encounter a number of host cell types, including pericytes, reactive glia, neural progenitor cells, neurons, and oligodendrocytes [57][161]. Once normal astrocytes encounter cancer cells, they become reactive astrocytes (RAs) and limit the survival of arriving metastatic cells at the initial stages of BCBM [58][162]. As a result, most cancer cells are unable to tolerate the neuroinflammatory reaction that is rapidly instigated by microglia and astrocytes [59][60][163,164]. Molecular profiling of paired brain metastases and corresponding primary breast tumors by whole-exome sequencing revealed that brain metastases harbored part of genomic aberrations in the PI3K/AKT/mTOR pathways, which were not detected in the corresponding primary tumor [31][121].
4. Therapeutic Implications of the Genomic Information
In the past decade, significant efforts were made to characterize the genetic drivers in BC metastasis using modern sequencing techniques. Better understanding of the genomic complexity and heterogeneity of BC metastasis will lead to improved treatment strategies and research directions. Moreover, the extent of response and potential resistance to treatments varies among different metastatic types and patients. Therefore, more effective and targeted treatments that build on the molecular mechanism of meta-static BC are needed.
Given that MTDH mediates the adhesion of cancer cells to lung endothelium, many ways against MTDH were developed to inhibit lung metastasis of BC, e.g., the antibodies reacting to the LHD of MTDH, the multiple tyrosine kinase inhibitor (TKI) SU6668, and DNA vaccines. In addition to molecules that mainly mediate single-organ metastasis, several molecules exhibit pleiotropic functions in metastasis to multiple organs. Actionable mutations in PI3K/AKT/mTOR pathway are frequent in bone, lung, live and brain metastasis of BC patients from above sequencing evidence. Overexpression of CCL2 promotes BC metastasis to both lung and bone in a manner de-pendent on its receptor CCR2 expressed on stromal cells to increase macrophage infiltration and osteoclast differentiation.
Moreover, metastasis is a multigenic process, establishing the functional role of candidate metastasis genes may not be easily accomplished by introducing individual genes into weakly metastatic cells to enhance their metastatic phenotype. MiRNAs, 20–22 short nucleotide sequences that often negatively regulate gene expression, can regulate multiple genes and hence multiple processes simultaneously. Since miRNA can target multiple sets of genes, it is an excellent clinical choice for cancer metastasis, a process mediated by multiple deregulated genes. Various drugs targeting tumor microenvironment components were already investigated in clinical trials, including antiangiogenic therapies, anti-inflammatory therapies, immunotherapies, and combination therapy [61][181].