Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Haiqiang Yao and Version 2 by Vivi Li.
The gut microbiota exists throughout the full life cycle of the human body, and it has been proven to have extensive impacts on health and disease. Accumulating evidence demonstrates that the interplay between gut microbiota and host epigenetics plays a multifaceted role in health maintenance and disease prevention. Intestinal microflora, along with their metabolites, could regulate multiple epigenetic pathways; e.g., DNA methylation, miRNA, or histone modification. Moreover, epigenetic factors can serve as mediators to coordinate gut microbiota within the host.
- gut microbiota
- SCFAs
- epigenetics
- microRNA
- interplay mechanism
1. Introduction
Microbiota are ecological communities of commensal microorganisms that typically inhabit a particular environment. The human microbiota, especially gut microbiota, affect host physiology and pathology to a great extent [1]. Human beings and microbiota are engaged in a dynamic equilibrium of interdependence and interplay. Microbes are distributed on the surface and in the external cavity of the human body, such as the oral cavity, the nasal cavity, and the intestinal lumen. The largest microbial community of microbiota in the human body is located in the large intestine; these obligate anaerobic organisms are known as gut microbiota [2]. Trillions of microorganisms live in the enteric canal, forming a complex symbiotic ecosystem with the host [3][4][3,4]. It is still unclear whether gut microbiota begin to colonize the human body before or after birth [5][6][5,6].
It should be noted that gut microbiota comprehensively affect a variety of physiological and pathological processes in the human body. According to the studies we could access to date, commensal gut microbiota play a vital role in maintaining the body’s homeostasis and the occurrence of disease [7]. Microorganisms in the gut are involved in many human physiological functions; e.g., fermentation of indigestible food components and synthesis of vitamins, resistance to viruses, maintenance of intestinal homeostasis, promotion of immune system maturation, and maintenance of intestinal epithelial barrier function [8]. Therefore, gut microbiota have even been considered to be an essential organ and called “the second genome”.
In recent years, with the rapid advance of gut microbiota research, it has been revealed that acquired factors such as diet, antibiotic use, infections, etc., could regulate microbial communities via epigenetic approaches, affecting the homeostasis and disorder status of a host [9]. Epigenetic modifications can affect a host’s pathological conditions by manufacturing a nutritional profile or shaping the structure of gut microbiota. For example, microRNA (miRNAs), as a minimal mediator, can enter the gut microbiota and shape their composition, ultimately affecting the host’s intestinal health [8][10][11][12][13][14][8,10,11,12,13,14].
The crosstalk between microbiota and epigenetics, a remarkable academic focal point, has been attracting more and more attention. Various important but obscure mechanisms are thought to have been uncovered from this perspective. Studies have shown that this interplay mechanism accompanies the entire life cycle of the human body, from the fetal period to the end of life, and has an important impact on health maintenance and disease prevention [8] (Figure 1). However, at present, the exact mechanism of the interplay between gut microbiota and epigenetics is only the tip of the iceberg.
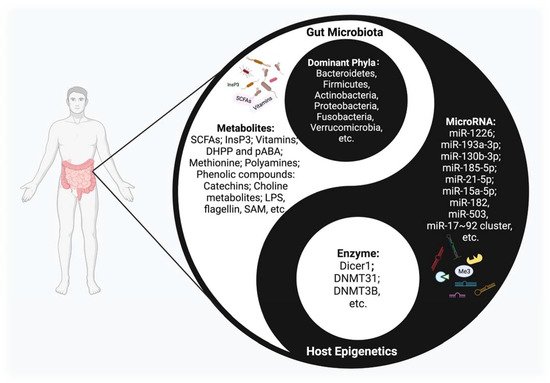
Figure 1. The Interplay between host epigenetic mechanisms and gut microbiota. Gut microbiota inhabit the intestinal tract and are in symbiosis harmoniously with the human body, constituting a microecology in a yin-yang dynamic balance. Once this balance is disrupted, various diseases may occur. The host regulates the diversity and composition of gut microbiota through miRNAs and other epigenetic factors. Intestinal flora, along with their metabolites, could regulate multiple epigenetic pathways; e.g., DNA methylation, miRNA, or histone modification in the host.
2. Overviews of Epigenetics and Gut Microbiota Research
2.1. Epigenetics
Epigenetics is the study of changes in organisms caused by the modification of gene expression rather than the alteration of a DNA sequence; meanwhile, phenotype changes can be stably transmitted in the process of growth and development [15][16][17,18]. Epigenetic mechanisms include DNA methylation, histone modification, and noncoding RNAs. These host epigenetic modifications play an intermediary role in intestinal homeostasis, inflammatory regulation, and metabolic disorders, and provide a basis for maintaining the intestinal commensal system.
2.1.1. DNA Methylation
DNA methylation is an important epigenetic regulatory mechanism in the human body that affects gene expression by regulating the accessibility of transcription factors [17][18][19,20]. The mechanism that allows DNA methylation sites to be retained during DNA replication is characteristic of epigenetic systems, and is a process that benefits from specific proteins that recognize CpG hemimethylation in DNA, thereby reproducing DNA methylation on newly replicated DNA [19][21]. DNA methylation is operated by the DNA methyltransferase DNMT1 and its assistant UHRF1 (also known as NP95), which specifically binds hemimethylated DNA by stimulating DNMT1 via its ubiquitin ligase. Therefore, gene complexes are tightly linked to the “author” and “reader” of epigenetic methyl CpG marks, two roles that are essential for maintaining DNA methylation [20][22]. This process needs numerous raw materials; dietary-derived nutrients such as folate and vitamin B12 could serve as methyl donors, and methyl groups catalyzed by DNA methyltransferase can be transferred to the basic group in the DNA sequence [21][23]. DNMT can add a methyl group from the donor S-adenosylmethionine (SAM) to the carbon-5 position of the cytosine (5mC), while the ten-eleven translocation enzyme (TET) dioxygenase family can actively reverse this process by oxidizing 5mC to 5-hydroxy-methylcytosine (5hmC) [17][19]. It is generally believed that there are four kinds of DNMT in mammals, and these can be divided into two categories: DNMT1 and DNMT3. DNMT1 maintains its methylation during DNA replication and repair, and DNMT3 catalyzes the de novo methylation of CPG [22][24]. Comparing the gut microbiota of GF and conventional mice, the degree of intestinal DNA methylation was significantly reduced in the absence of gut microbiota [23][25]. This hypomethylation is not caused by a low level of DNMT activity; however, the underlying reason is supposed to be the reduction of one-carbon metabolites derived from gut microbiota [24][26].
2.1.2. Histone Modification
Histones are important components of chromatin, and they fold DNA to assemble nucleosome structures consisting of H2A, H2B, H3, and H4. Histone modification, as an important epigenetic pathway, plays an important role in regulating gene replication, transcription, and DNA damage repair. Multiple modifications can be made to histones, including acetylation, methylation, phosphorylation, and ubiquitination, primarily in the N-terminal histone tails, and most of the histone modifications are reversible and responsive to metabolic changes [18][20].
Histone acetylation, an important type of histone modification, is generally related to active gene transcription, while deacetylation is related to the inhibition of transcription [18][20]. The status of histone acetylation is regulated by both histone acetyltransferases (HATs) and histone deacetylases (HDACs), which play opposite roles. The biofunction of HATs is to catalyze the deposition of acetyl group by transferring an acetyl group from acetyl-coenzyme A to the ε-amino group of lysine residues. Histone acetylation can also expose the target sites of transcription factors in nucleosome DNA and initiate the assembly of transcription complexes. Contrary to the functions of HATs, HDACs catalyze deacetylation to erase the acetyl group from the tail of histone leading to a reduction of accessibility.
Histone methylation is catalyzed by a large number of histone methyltransferases. There are multiple forms of histone methylation, depending on which amino acids in the histones are methylated and how many methyl groups are attached. The most common form is the methylation of lysine residue ε-amino. Histone methylation can regulate the transcription of genes, including both promotion and inhibition, under different conditions [25][27]. Histone methyltransferases (HMT) have different specificities for histone substrates, but all known HMTs use S-adenosylmethionine (SAM, also known as AdoMet) as a methyl donor and produce S-adenosylhomocysteine (SAH, also known as AdoHcy) as a byproduct. SAH is a competitive inhibitor of SAM and a noncompetitive or mixed peptide substrate inhibitor [25][27].
There are several ways in which gut microbiota can regulate histone modifications. Generating numerous bioactive compounds from microbial metabolism is one effective mechanism. Acetate and propionate can inhibit HDAC2 and HDAC3, and butyrate can inhibit the activity of HDAC1 and HDAC2, thereby affecting the stability of intestinal ecology [26][27][28,29]. Meanwhile, histone methylation or demethylation can be globally modulated by various cellular metabolites/cofactors, including SAM, Fe2+/Fe3+, and α-KG [28][30].
2.1.3. Noncoding RNA
The discovery of noncoding RNAs is considered a scientific breakthrough in the “genetic central dogma” (DNA→RNA→Protein) and the “junk RNA” theory, in which the junk RNA contains all those RNAs that are not translated—about 70% of the genome is transcribed, while about only 2% is translated—and, therefore, they are considered non-functional [29][31]. Noncoding RNAs participate in important biological processes, but do not encode proteins. Thus far, these molecules have been widely demonstrated to play a role in the regulation of the translation and transcription of coding and noncoding genes. Based on their sizes, regulatory noncoding RNAs can be divided into long ncRNAs (lncRNAs; >200–300 bp) and small ncRNAs (<200–300 bp), including miRNAs; short interfering RNAs (siRNAs); endo-siRNAs; and PIWI-interacting RNAs (piRNAs) [30][32]. Noncoding RNAs have many important functions, including (1) structural functions, such as the construction of ribosomal subunits (rRNAs); (2) transport functions during protein translation, in which tRNAs interact with RNA ligands and mRNAs; and (3) regulatory functions by regulating RNA, DNA, and proteins [31][33].
In particular, lncRNAs have been reported to be useful for distinguishing types of gut microbiota and as biomarkers for identifying the process of host–microbiota interplay [32][34]. The expression of lncRNAs in mice with or without the presence of microflora are revealed to be distinct. Furthermore, the different strains of microbes may also affect the lncRNA expression. Mice inoculated with wild-type E. coli and E. coli expressing bile salts hydrolase induced specific changes of lncRNA profiles [32][34].
MiRNA, an endogenous small noncoding RNA molecule, contains about 18 to 25 nucleotides and can regulate gene expression by inducing degradation of mRNAs or inhibiting translation. Meanwhile, miRNA can regulate various physiological or pathological pathways such as cell differentiation, cell proliferation, and tumor development; approximately 30% of protein-encoding genes are regulated by miRNAs [33][35]. MiRNAs also can regulate the commensal microbiota-dependent intestinal epithelial cells that maintain gut homeostasis and dysbiosis [34][36]. Meanwhile, miRNA is an important regulator of the immune pathway that is involved in homeostasis. The dysfunction of miRNAs causes a wide variety of body abnormalities, including cancer and autoimmune diseases [35][37].
2.2. Gut Microbiota
In human gut microbiota, the dominant phyla are Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, Fusobacteria, and Verrucomicrobia, two of which—Firmicutes and Bacteroidetes—account for 90% of gut microbiota [2][36][2,38]. Although mature microbiota are rather stable, their richness and composition may fluctuate with internal and external factors, including age, region, lifestyle, drug use, and dietary habits. Gut microbiota are present on the surface of the intestinal mucosa and vary according to the different anatomical regions of the gut; they play an essential role in digesting food, obtaining energy, and maintaining homeostasis [36][37][38,39].
In particular, gut microbiota generate numerous bioactive compounds, including short-chain fatty acids (SCFAs) and choline metabolites and lipids [37][39], which are important factors affecting host physiology and pathology [8]. Microbial metabolites are key messengers in the interplay between microbiota and epigenetics. They not only produce local effects in the gut, but also regulate distant organs, such as the lungs, the heart, and the brain [38][40]. The dysbiosis of gut microbiota can induce a series of diseases, such as obesity, diabetes, metabolic syndrome, and inflammatory bowel disease (IBD) [39][40][41][42][41,42,43,44]. The microbial community plays a key role in maintaining the balance of the intestinal environment.
Until recently, research has mainly focused on exploring the occurrence, development, and mechanism of disease from the perspective of gut microbiota. However, the interplay between intestinal microorganisms and epigenetics is receiving increased attention, and multiple studies have demonstrated the significance of this mechanism in health maintenance and disease prevention.
3. The Effects of Epigenetic Regulation on Gut Microbiota
3.1. Modulation of the Gut Microbiota Composition
Epigenetic mechanisms can influence the microbial community and microbiota-derived metabolites; specifically, miRNA is deeply involved in microbiota shaping [43][45]. Recent studies have shown that host miRNAs are involved in the regulation of gut microbiota. MiRNAs in the intestinal lumen are derived from intestinal epithelial cells (IECs), as well as goblet and paneth cells, and are excreted through exosomes [44][46]. MiRNAs from the host can specifically regulate the transcription of microbial genes and then affect the growth of intestinal microflora and microbiota structure [45][47]. A multitude of studies have confirmed that miRNAs can control the growth of microbiota and shape the structure of the microbial community [46][47][48,49]. For example, coculture of the miRNAs with microbiota can induce significant gene expression changes in microbiota [44][46]. By adding synthesized miRNA to the bacterial culture medium in vitro, we found that miRNA changed the gene expression of bacteria, such as F. nucleatum and E. coli. Furthermore, mice given specific miRNA also demonstrated changes in the E. coli growth and bacterial gene transcripts. The results indicated the levels of miRNA were negatively correlated with the abundance of the gut microbiota [45][47] (Table 1).
3.2. Adjustment of the Intestinal Homeostasis
Host epigenetics, especially miRNAs, can participate in the physiological functions related to maintaining intestinal homeostasis by regulating gut microbiota. For example, the expression of miR-21-5p in IECs can regulate intestinal epithelial permeability through ADP ribosylation factor 4 (ARF4) [48][50]. Studies found that the loss of host miRNAs in feces, or the selective knockout of miRNA-processing enzymes in intestinal epithelium, may lead to an imbalance of microbial homeostasis and an aggravation of colitis. These phenomena demonstrate that host-secreted miRNAs could send feedback to the gut microbiota to maintain intestinal homeostasis.
Gut microbiota, along with the intestinal epithelium and mucosal system, establish a complex intestinal barrier against enteropathogens [49][51]. A variety of epigenetic mechanisms are involved in the process of intestinal barrier formation through regulating the differentiation of IECs, as well as the richness of gut microbiota and their derived metabolites [50][52]. Host epigenetic effects vary between different types of gut microbiota. Related studies suggest that host-secreted miRNAs have an important impact on intestinal homeostasis by regulating the growth and structure of microbial communities, providing a new perspective for maintaining intestinal health [46][48]. However, the detailed mechanism of how these epigenetic factors interplay with different cues from symbiotic microbiota still appears foggy, and further investigation into this topic is needed in the future.
3.3. Regulation of the Host Metabolism
Gut microbiota and their metabolites have been identified as effective metabolic regulators, and epigenetic mechanisms could influence the host metabolism via this pathway [46][48] (Figure 2). The latest studies show that the interplay between circulating miRNA and gut microbiota serves as a notable mechanism in obesity [51][53]. Compared with the control group, 26 different circulating miRNAs and 12 microbial species were screened in an obese group, and these target miRNAs and microbes were significantly related. Three miRNAs (miR-130b-3p, miR-185-5p, and miR-21-5p) showed a negative correlation with Bacteroides eggerthii and exerted a BMI-regulating function. Furthermore, the expressions of miR-107, miR-103a-3p, miR-222-3p, and miR-142-5p were negatively associated with B. intestinihominis abundance. These miRNAs regulate genes involved in metabolism-related pathways, including fatty-acid degradation, insulin signaling, and glycerol lipid metabolism. MiR-15a promotes insulin biosynthesis by inhibiting the expression of endogenous uncoupling protein 2 (UCP2), increasing the level of ATP and glucose-stimulated insulin secretion. The expression of miR-15a-5p was negatively correlated with the abundance of H. parainfluenzae and could affect insulin levels, as was demonstrated by in vivo data. Meanwhile, a bioinformatic analysis showed that the expression levels of 14 circulating miRNAs (miR-107, miR-103a-3p, miR-142-5p, miR-222-3p, miR-221-3p, miR-183-5p, miR-130b-3p, miR-15a-5p, miR-33a-5p, miR-210-3p, miR-144-3p, miR-185-5p, miR-130a-3p, and miR-21-5p) and the richness of four intestinal microflora taxa (D. longicatena, B. intestinihominis, B. eggerthii, and H. parainfluenzae) in the obese group were significantly different; a correlation analysis indicated that there is an interplay mechanism between miRNAs and microbes [51][53]. The result indicated that miRNAs play a role in communication between the gut microbiota and the host, and that the crosstalk between epigenetics and gut microbiota is a potential target for treating metabolic disorders; however, further investigation is needed to uncover the mechanism more deeply.
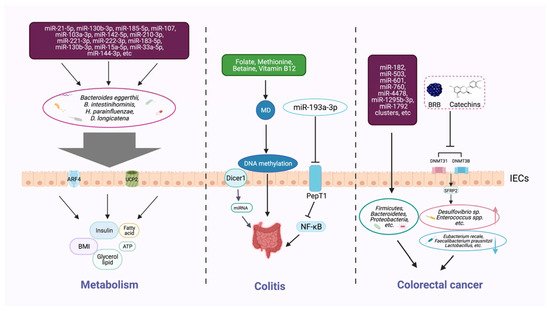
Figure 2. Host epigenetic factors regulate the gut microbiota in multiple progresses of diseases. The gut epithelium secretes a variety of miRNAs, which can enter microorganisms to affect their transcription and alter the microbial structure and diversity. The gut microbiota generates different metabolites (e.g., butyrate and bile acids) that can, in turn, regulate the host metabolism, including BMI, insulin secretion, and lipid production. Dietary intake of nutrients (e.g., folic acid, methionine, and vitamin B12) provides methyl donors that affect host DNA methylation, which may modulate the intestinal inflammatory state. Some microbial metabolites can modify DNA methylation, histone acetylation, and miRNAs, and impair the homeostasis of the intestinal environment, thereby reducing beneficial microbiota and increasing the richness of pathogenic bacteria, and inducing the development of colorectal cancer.
3.4. Factors Involved in the Progress of Colitis
Epigenetic factors that arise through manipulating gut microbiota can affect the occurrence and development of colitis (Figure 2). Epigenetic-related “writer” and “eraser” enzymes play a crucial role in preventing IBD. For example, when intestinal epithelial cells lack the enzyme Dicer1, which is essential for miRNA processing, the expression level of miRNA in gut contents and feces will be reduced, resulting in gut microbiota imbalance and severe colitis [44][46]. Furthermore, fecal miRNA transplantation recovers the structure of gut microbiota and improves IBD. These findings indicate that miRNA plays a physiological role in shaping gut microbiota and improving intestinal inflammation. Another study found that miRNA-193a-3p indirectly reduces microbiota-metabolite-induced colon inflammation. PepT1 is a transporter that can absorb microbial products. PepT1 was found to be upregulated and negatively correlated with mir-193a-3p in ulcerative colitis (UC). Further verification showed that mir-193a-3p decreased the activity and expression of target gene PepT1, and subsequently reduced the uptake of microbial products and inhibited the NF-κB pathway. These results suggest that miR-193a-3p can mitigate the intestinal inflammation caused by microbial products and protect intestinal homeostasis [52][54] (Table 1).
Table 1. Mechanisms of epigenetic modifications that regulate gut microbiota.
| Epigenetic Factors | Epigenetic-Associated Gut Microbiota | Mechanism | Effects on Health or Diseases | Reference |
|---|---|---|---|---|
| MicroRNA-1226 | Escherichia coli | Enter the interior of strains | Regulate microbial gene transcripts and affect microbial growth | [45][47] |
| MicroRNA-193a-3p | Gut microbiota metabolism | Downregulate PepT1 and then suppress the NF-κB pathway by reducing the intake of gut microbiota metabolism in the colon | Reduce inflammatory in the colon, protect gut homeostasis |
[52][54] |
| MiR-130b-3p, MiR-185-5p, MiR-21-5p | Bacteroides eggerthi | Negatively regulate the expression of genes | BMI and host metabolism pathways (fatty-acid degradation, insulin signaling, glycerol lipid metabolism) | [51][53] |
| MicroR-15a-5p | H. parainfluenzae | Negative correlation | Inhibit the expression of endogenous UCP2; increase the level of ATP, and stimulate insulin secretion | [51][53] |
| MicroRNA enzyme Dicer1 | Structure of gut microbiota | Reduce the level of miRNA | The absence of Dicer1 may result in dysbiosis of gut microbiota and aggravation of colitis | [44][46] |
| 76 microRNAs (including miR-182, miR-503, and miR-17~92 cluster, etc.) | Firmicutes, Bacteroidetes, and Proteobacteria | A mediating role in which they transfer to stromal cells and immune cells if miRNAs in tumor cells are out of balance | Affect tumor metastasis and invasion | [53][55] |
| DNMT31 and DNMT3B promote the methylation of SFRP2 promoter | Desulfovibrio sp. and Enterococcus spp.; Eubacterium rectale, Faecalibacterium prausnitzii and Lactobacillus | Decrease of Desulfovibrio sp. and Enterococcus spp.; Increase of Eubacterium rectale, Faecalibacterium prausnitzii, and Lactobacillus |
Increase intestinal inflammation and risk of colon cancer | [54][56] |
Methyl-donors (MD), including many dietary supplements, such as folate, methionine, betaine, and vitamin B12, can provide essential raw material in DNA methylation processes [55][57]. The colon is an important environment for the folate metabolic cycle to maintain local homeostasis [56][58]. In a mouse model of Crohn’s disease (CD), methyl-donor supplementation can prevent the intestinal colonization of adherent-invasive E. coli, showing beneficial effects on the inflammation. Furthermore, the serum folate concentration was found to be inversely correlated to fecal inflammatory markers in a cohort of CD [57][59]. MD supplementation during pregnancy increased the odds of colitis infection in offspring, and was found to be associated with persistent epigenetic and intestinal microbial changes [55][57]. Methionine is an active MD, and short-term dietary methionine supplementation in a mouse model may alter the DNA methylation in the gut; regulate the microbiome profiles; and affect the intestinal barrier function, gene expression, and histomorphology [58][60].
3.5. Factors Associated with Colorectal Cancer
Epigenetic changes in the colon can markedly help us identify and prevent intestinal tumors (Figure 2). For instance, changes in miRNAs have been observed to affect the state of gut tumor metastasis and invasion. Accumulating evidence confirmed that miRNAs are involved in the occurrence, development, and response to treatment of tumors, and can be used as biomarkers for diagnosis, prognosis, and prediction of therapeutic effects [48][59][50,61]. Compared with healthy controls, the plasma levels of miR-601 and miR-760 in colorectal cancer (CRC) patients were significantly lower, which is helpful for the early diagnosis of CRC [60][62]. Moreover, lower expression levels of mir-4478 and mir-1295b-3p in fecal samples could also serve as potential noninvasive molecular markers for CRC diagnosis [61][63].
Further investigation revealed that the specific miRNAs in CRC are closely linked with microorganisms [53][55]. A total of 76 miRNAs, including the known miR-182, miR-503, and miR-1792 clusters, were differently expressed in tumor tissues than in healthy tissues in CRC; those miRNAs were associated with the presence of several phyla of microbes, such as Firmicutes, Bacteroidetes, and Proteobacteria. The imbalance of miRNA in tumor cells may affect the survival of gut microbiota or regulate the gene expression of certain microbes [53][55] (Table 1). Studies have confirmed that the microbial community relies on miRNAs that play a mediating role in maintaining crosstalk with the host [62][63][64,65].
Microarray analysis identified genes with different DNA methylation in colon cancer [64][66]. According to another study, black raspberry BRB anthocyanins could downregulate the expression of DNMT3A, DNMT3B, and p-STAT3 in CRC, resulting in the demethylation of the SFRP2 gene promoter and increasing the expression of SFRP2 at both the mRNA and protein levels. Along with this epigenetic change, the microbial structure has also been modified, which implies an inner correlation of DNA methylation and gut microbiota. The interplay of the two factors is supposed to play a crucial role in the prevention of CRC [54][56].