This review discusses molecular adducts, whose composition allows a symmetric structure. Such adducts are popular model systems, as they are useful for analyzing the effect of structure on the property selected for study since they allow one to reduce the number of parameters. The main objectives of this discussion are to evaluate the influence of the surroundings on the symmetry of these adducts, steric hindrances within the adducts, competition between different noncovalent interactions responsible for stabilizing the adducts, and experimental methods that can be used to study the symmetry at different time scales. This review considers the following central binding units: hydrogen (proton), halogen (anion), metal (cation), water (hydrogen peroxide).
- hydrogen bonding
- noncovalent interactions
- isotope effect
- cooperativity
- water
- organometallic complexes
- NMR
- DFT
1. Introduction
If something is perfectly symmetric, it can be boring, but it cannot be wrong. If something is asymmetric, it has potential to be questioned. Note, for example, the symmetry of time in physics [1,2][1][2]. Symmetry also plays an important role in chemistry. Whether it is stereochemistry [3], soft matter self-assembly [4,5][4][5], solids [6[6][7],7], or diffusion [8], the dependence of the physical and chemical properties of a molecular system on its symmetry is often a key issue. Symmetric molecular adducts are popular model systems; they are used to analyze the effect of structure on the property chosen for research since they allow one to reduce the number of parameters [9,10,11,12][9][10][11][12]. On the other hand, symmetry in chemistry is a matter of the size and time scale in question [13]. The same molecular system can be symmetric for one experimental method and asymmetric for another. It is important to understand what processes are hidden behind this discrepancy in each specific case.
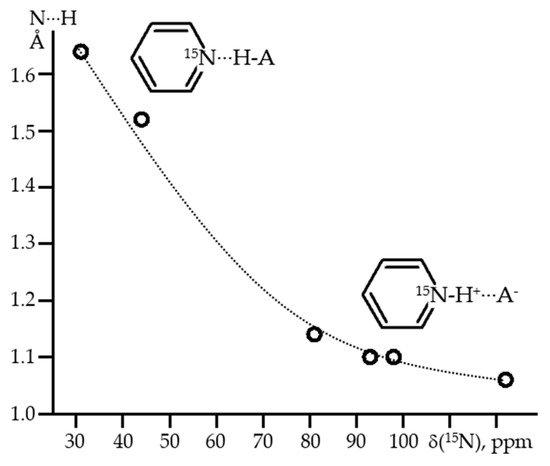
The problem of the size scale already begins at the level of the model adducts composition. What structure has the simplest model adduct with which it is possible to investigate the property under consideration? The Zundel cation (H5O2+) and the Eigen cation (H9O4+) seem to be the most illustrative example [14,15][14][15]. Which of these two structures is the best for simulating a hydrated proton? It seems that neither experiment nor theory can answer this question regardless of the property being discussed [16,17,18,19,20][16][17][18][19][20]. The same is valid for the hydration of the hydroxide ion [21,22,23,24][21][22][23][24]. Of course, bulk water is one of the most complex solvents in this content. The time scale problem has to do with tautomerism. For some methods, its rate is slow. In this case, experimental parameters can be observed for each of the structures presented. For other methods, this rate is fast and only average experimental parameters can be observed.
2. Hydrogen (Proton) as the Binding Unit
2.1. Intramolecular H-Bonds
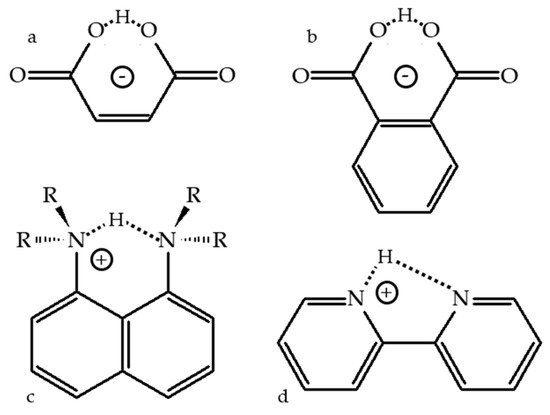
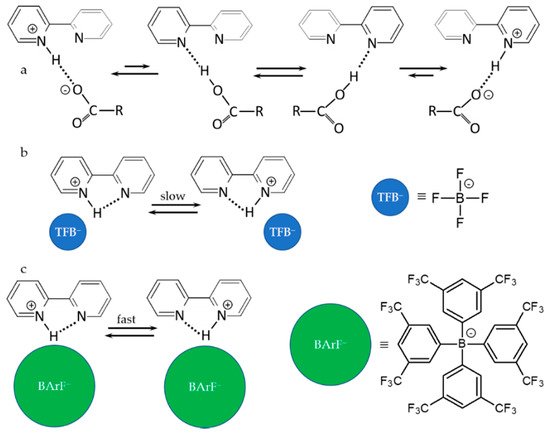
2.2. Proton-Bound Homodimers
Proton-Bound Homodimers
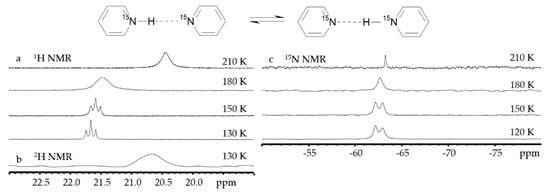
3. Halogen (Anion) as the Binding Unit
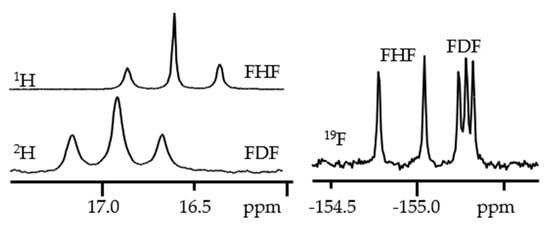
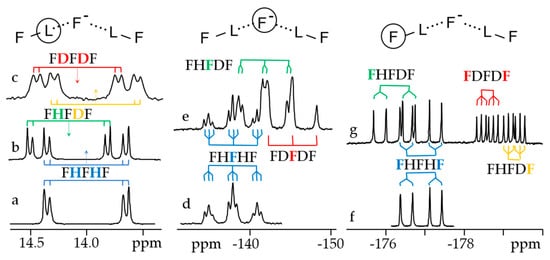
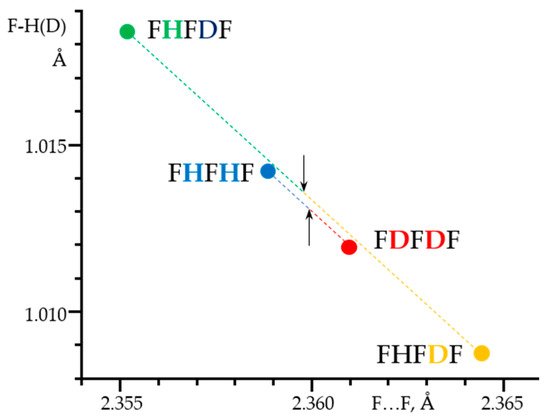
Figure 7 shows 1H, 2H and 19F NMR spectra of solutions containing FH···F-···HF, FH···F-···DF and FD···F-···DF anions in CDF3/CDF2Cl at 130 K [203][89]. The protons of FH···F-···HF are located at the outer fluorine atoms. The corresponding spin–spin scalar coupling 1JHF = 354 Hz. The protons also couple to the central fluorine atom across the H-bonds, hJH···F = −24 Hz. Therefore, these protons give rise to a doublet of doublet signal, Figure 7a. The outer and central fluorine atoms couple to each other, 2hJF…F = 147 Hz, and give rise to a doublet of doublet signal, Figure 7f, and a triplet of triplet signal, Figure 7d. The rate constant for proton and H-bond exchange is less than 103 s−1. This complex is not linear, the FFF angle is about 130° [191][90]. There is an anti-cooperative coupling of these two H-bonds. As a result, the FH···F-···DF anion is asymmetric. The 1H NMR chemical shift of the FH···F- proton is larger than the 2H NMR chemical shift of the F-···DF deuteron. The former is also larger, and the latter is less than the 1H NMR chemical shift of the protons in FH···F-···HF (see the arrows in Figure 7b,c). Consequently, the FH···F- H-bond is shorter and the F-···DF one longer than the bonds in FH···F-···HF. This conclusion is confirmed by changes of the coupling constants and 19F NMR chemical shifts. For example, in FH···F-···DF, for the FH···F- H-bond 1JHF = 348 Hz, hJH···F = −22 Hz and 2hJF…F = 151 Hz, while for the F-···DF H-bond 2hJF…F = 140 Hz.
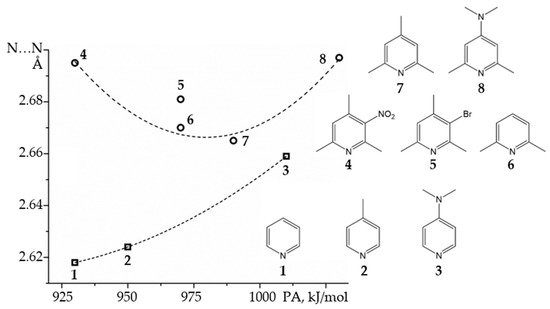
4. Metal (Cation) as the Binding Unit
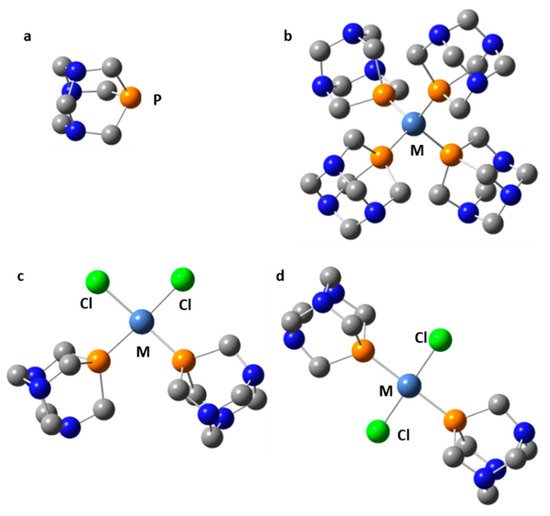
Symmetric transition metal organometallics are not necessarily the most effective catalysts. However, the elucidation of their structure in solution can be greatly facilitated if they are or can be symmetric. Although 1H and 13C NMR are not always sufficient to determine the structure of organometallic species, 31P NMR can be very useful when phosphorous is coordinated to the metal center. Only 1,3,5-triaza7-phosphaadamantane (PTA, Figure 9) complexes will be considered here. The rationale for this choice is explained as follows. The 31P isotope is the only stable isotope of phosphorus. It has a spin quantum number of 1/2 and a wide chemical shift range of about 400 ppm. This nucleus is a very convenient NMR probe for studying molecular complexes [99[58][91][92][93],204,205,206], organometallics [207,208[94][95][96][97],209,210], and mobility at interfaces [211,212,213][98][99][100]. For example, 31P NMR has been used to study the effect of temperature and hydration on the mobility of small to bulky molecules loaded onto mesoporous silica [214][101]. However, 31P NMR shielding can depend on the conformation of the molecule [215][102], the crystalline electric field [216][103], while various noncovalent interactions can cause similar changes [217][104].
5. Water (Hydrogen Peroxide) as the Binding Unit
6. Conclusions
- (i)
-
Steric hindrance and structural rigidity are not the only reasons why complexes can be asymmetric in the gas phase.
- (ii)
-
At any given moment of time, the solvation shell is somewhat asymmetric.
- (iii)
-
Dynamic processes in crystalline solids can be facilitated if the initial and final states are equivalent.
- (iv)
-
The motion of the proton in a H-bond should always be treated as quantum.The reader may find it useful to refer to other recent publications on the interactions of tetrahedral pnicogen and tetrel centres with Lewis bases [248][106], the coordination of triel centers [249][107], tetraphosphido complexes [250][108], dinuclear metal hydride complexes [251][109], crystalline peroxosolvates [252][110], the self-association of phosphonic acids [253][111], intramolecular H-bond dynamics [254][112], and a consistent description of noncovalent interactions [255][113].
References
- Mehlberg, H. Time, Causality, and the Quantum Theory. In Studies in the Philosophy of Science; Fawcett, C.R., Cohen, R.S., Eds.; Springer: Berlin/Heidelberg, Germany, 1980.
- Leifer, M.S.; Pusey, M.F. Is a time symmetric interpretation of quantum theory possible without retrocausality? Proc. R. Soc. A 2017, 473, 20160607.
- Castilla, A.M.; Ramsay, W.J.; Nitschke, J.R. Stereochemistry in Subcomponent Self-Assembly. Acc. Chem. Res. 2014, 47, 2063–2073.
- Wang, Y.-L.; Li, B.; Sarman, S.; Mocci, F.; Lu, Z.-Y.; Yuan, J.; Laaksonen, A.; Fayer, M.D. Microstructural and Dynamical Heterogeneities in Ionic Liquids. Chem. Rev. 2020, 120, 5798–5877.
- Wang, Y.; Wang, Y.; Breed, D.; Manoharan, V.N.; Feng, L.; Hollingsworth, A.D.; Weck, M.; Pineet, D.J. Colloids with valence and specific directional bonding. Nature 2012, 491, 51–55.
- Ungur, L.; Le Roy, J.J.; Korobkov, I.; Murugesu, M.; Chibotaru, L.F. Fine-tuning the Local Symmetry to Attain Record Blocking Temperature and Magnetic Remanence in a Single-Ion Magnet. Angew. Chem. Int. Ed. 2014, 53, 4413–4417.
- Sun, W.-B.; Yan, P.-F.; Jiang, S.-D.; Wang, B.-W.; Zhang, Y.-Q.; Li, H.-F.; Chen, P.; Wang, Z.-M.; Gao, S. High symmetry or low symmetry, that is the question—high performance Dy(iii) single-ion magnets by electrostatic potential design. Chem. Sci. 2016, 7, 684–691.
- Peng, J.; Cao, D.; He, Z.; Guo, J.; Hapala, P.; Ma, R.; Cheng, B.; Chen, J.; Xie, W.J.; Li, X.-Z.; et al. The effect of hydration number on the interfacial transport of sodium ions. Nature 2018, 557, 701–705.
- Shenderovich, I.G. 1,3,5-Triaza-7-Phosphaadamantane (PTA) as a 31P NMR Probe for Organometallic Transition Metal Complexes in Solution. Molecules 2021, 26, 1390.
- Grabowski, S.J. Hydrogen Bond and Other Lewis Acid–Lewis Base Interactions as Preliminary Stages of Chemical Reactions. Molecules 2020, 25, 4668.
- Grabowski, S.J.; Ugalde, J.M.; Andrada, D.M.; Frenking, G. Comparison of Hydrogen and Gold Bonding in [XHX]-, [XAuX]-, and Isoelectronic [NgHNg]+, [NgAuNg]+ (X=Halogen, Ng=Noble Gas). Chem. Eur. J. 2016, 22, 11317–11328.
- Alkorta, I.; Elguero, J. Review on DFT and ab initio Calculations of Scalar Coupling Constants. Int. J. Mol. Sci. 2003, 4, 64–92.
- Kellman, M.E. Symmetry in chemistry from the hydrogen atom to proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 14287–14294.
- Zundel, G.; Metzger, H. Energiebänder der tunnelnden Überschuß-Protonen in flüssigen Säuren. Eine IR-spektroskopische Untersuchung der Natur der Gruppierungen H5O2+. Z. Phys. Chem. 1968, 58, 225–245.
- Wicke, E.; Eigen, M.; Ackermann, T. Über den Zustand des Protons (Hydroniumions) in wäßriger Lösung. Z. Phys. Chem. 1954, 1, 340–364.
- Carpenter, W.B.; Yu, Q.; Hack, J.H.; Dereka, B.; Bowman, J.M.; Tokmakoff, A. Decoding the 2D IR spectrum of the aqueous proton with high-level VSCF/VCI calculations. J. Chem. Phys. 2020, 153, 124506.
- Wang, E.; Zhu, B.; Gao, Y. Discontinuous transition between Zundel and Eigen for H5O2+. Chin. Phys. B 2020, 29, 083101.
- Li, C.; Swanson, J.M.J. Understanding and Tracking the Excess Proton in Ab Initio Simulations; Insights from IR Spectra. J. Phys. Chem. B 2020, 124, 5696–5708.
- Vener, M.V.; Kong, S.; Levina, A.A.; Shenderovich, I.G. Spectroscopic Signatures of [H9O4]+ and [H13O6]+ Ions in a Polar Aprotic Environment Revealed under DFT-PCM Approximation. Acta Chim. Slov. 2011, 58, 402–410.
- Vener, M.V.; Librovich, N.B. The structure and vibrational spectra of proton hydrates: H5O2+ as a simplest stable ion. Int. Rev. Phys. Chem. 2009, 28, 407–434.
- Shi, H.; Gong, L.-D.; Liu, C.; Lu, L.-N.; Yang, Z.-Z. ABEEM/MM OH- Models for OH-(H2O)n Clusters and Aqueous OH-: Structures, Charge Distributions, and Binding Energies. J. Phys. Chem. A 2020, 124, 5963–5978.
- Herbert, J.M. Fantasy versus reality in fragment-based quantum chemistry. J. Chem. Phys. 2019, 151, 170901.
- Vener, M.V.; Shenderovich, I.G.; Rykounov, A.A. A qualitative study of the effect of a counterion and polar environment on the structure and spectroscopic signatures of a hydrated hydroxyl anion. Theor. Chem. Acc. 2013, 132, 1361.
- Librovich, N.B.; Sakun, V.P.; Sokolov, N.D. H+ and OH- ions in aqueous solutions vibrational spectra of hydrates. Chem. Phys. 1979, 39, 351–366.
- Woo, H.-K.; Wang, X.-B.; Wang, L.-S.; Lau, K.-C. Probing the Low-Barrier Hydrogen Bond in Hydrogen Maleate in the Gas Phase: A Photoelectron Spectroscopy and ab Initio Study. J. Phys. Chem. A 2005, 109, 10633–10637.
- Garcia-Viloca, M.; González-Lafont, A.; Lluch, J.M. Theoretical Study of the Low-Barrier Hydrogen Bond in the Hydrogen Maleate Anion in the Gas Phase. Comparison with Normal Hydrogen Bonds. J. Am. Chem. Soc. 1997, 119, 1081–1086.
- Kiefer, P.M.; Hynes, J.T. Theoretical aspects of tunneling protontransfer reactions in a polar environment. J. Phys. Org. Chem. 2010, 23, 632–646.
- Limbach, H.-H.; Tolstoy, P.M.; Pérez-Hernández, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO Hydrogen Bond Geometries and NMR Chemical Shifts: From Equilibrium Structures to Geometric H/D Isotope Effects, with Applications for Water, Protonated Water, and Compressed Ice. Isr. J. Chem. 2009, 49, 199–216.
- Vener, M.V. Model study of the primary H/D isotope effects on the NMR chemical shift in strong hydrogen-bonded systems. Chem. Phys. 1992, 166, 311–316.
- Schah-Mohammedi, P.; Shenderovich, I.G.; Detering, C.; Limbach, H.-H.; Tolstoy, P.M.; Smirnov, S.N.; Denisov, G.S.; Golubev, N.S. H/D-Isotope Effects on NMR Chemical Shifts and Symmetry of Homoconjugated Hydrogen-Bonded Ions in Polar Solution. J. Am. Chem. Soc. 2000, 122, 12878–12879.
- Shenderovich, I.G.; Burtsev, I.G.; Denisov, G.S.; Golubev, N.S.; Limbach, H.-H. Influence of the Temperature-Dependent Dielectric Constant on the H/D Isotope Effects on the NMR Chemical Shifts and the Hydrogen Bond Geometry of Collidine-HF Complex in CDF3/CDClF2 Solution. Magn. Reson. Chem. 2001, 39, S91–S99.
- Gunnarsson, G.; Wennerström, H.; Egan, W.; Forsén, S. Proton and deuterium NMR of hydrogen bonds: Relationship between isotope effects and the hydrogen bond potential. Chem. Phys. Lett. 1976, 38, 96–99.
- Perrin, C.L.; Nielson, J.B. Asymmetry of Hydrogen Bonds in Solutions of Monoanions of Dicarboxylic Acids. J. Am. Chem. Soc. 1997, 119, 12734–12741.
- Perrin, C.L. Are Short, Low-Barrier Hydrogen Bonds Unusually Strong? Acc. Chem. Res. 2010, 43, 1550–1557.
- Mavri, J.; Hodošček, M.; Hadži, D. Ab initio SCF and Møller-Plesset calculations on the hydrogen bond in hydrogen malonate: Effects of neighbour ions and polarizable medium. J. Mol. Struct. 1990, 209, 421–431.
- Mavri, J.; Hadži, D. Influence of solvation on the hydrogen bond in hydrogen malonate an ab initio and semiempirical study. J. Mol. Struct. Theor. Chem. 1998, 432, 257–262.
- Shenderovich, I.G. Solid State NMR for Nonexperts: An overview of simple but general practical methods. Solids 2021, 2, 139–154.
- Golubev, N.S.; Detering, C.; Smirnov, S.N.; Shenderovich, I.G.; Denisov, G.S.; Limbach, H.-H.; Tolstoy, P.M. H/D Isotope Effects on NMR Chemical Shifts of Nuclei Involved in a Hydrogen Bridge of Hydrogen Isocyanide Complexes with Fluoride Anion. Phys. Chem. Chem. Phys. 2009, 11, 5154–5159.
- Golubev, N.S.; Melikova, S.M.; Shchepkin, D.N.; Shenderovich, I.G.; Tolstoy, P.M.; Denisov, G.S. Interpretation of H/D Isotope Effects on NMR Chemical Shifts of [FHF]- Ion Based on Calculations of Nuclear Magnetic Shielding Tensor Surface. Z. Phys. Chem. 2003, 217, 1549–1563.
- Di Muzio, S.; Ramondo, F.; Gontrani, L.; Ferella, F.; Nardone, M.; Benassi, P. Choline Hydrogen Dicarboxylate Ionic Liquids by X-ray Scattering, Vibrational Spectroscopy and Molecular Dynamics: H-Fumarate and H-Maleate and Their Conformations. Molecules 2020, 25, 4990.
- Shenderovich, I.G. Simplified Calculation Approaches Designed to Reproduce the Geometry of Hydrogen Bonds in Molecular Complexes in Aprotic Solvents. J. Chem. Phys. 2018, 148, 124313.
- Guo, J.; Tolstoy, P.M.; Koeppe, B.; Denisov, G.S.; Limbach, H.-H. NMR Study of Conformational Exchange and Double-Well Proton Potential in Intramolecular Hydrogen Bonds in Monoanions of Succinic Acid and Derivatives. J. Phys. Chem. A 2011, 115, 9828–9836.
- Garcia-Viloca, M.; González-Lafont, A.; Lluch, J.M. Asymmetry of the Hydrogen Bond of Hydrogen Phthalate Anion in Solution. A QM/MM Study. J. Am. Chem. Soc. 1999, 121, 9198–9207.
- Perrin, C.L.; Lau, J.S. Hydrogen-Bond Symmetry in Zwitterionic Phthalate Anions: Symmetry Breaking by Solvation. J. Am. Chem. Soc. 2006, 128, 11820–11824.
- Perrin, C.L. Symmetry of hydrogen bonds in solution. Pure Appl. Chem. 2009, 81, 571–583.
- Mori, Y.; Masuda, Y. Effect of solvent on proton location and dynamic behavior in short intramolecular hydrogen bonds studied by molecular dynamics simulations and NMR experiments. Chem. Phys. 2015, 458, 18–29.
- Piękoś, P.; Jezierska, A.; Panek, J.J.; Goremychkin, E.A.; Pozharskii, A.F.; Antonov, A.S.; Tolstoy, P.M.; Filarowski, A. Symmetry/Asymmetry of the NHN Hydrogen Bond in Protonated 1,8-Bis(dimethylamino)naphthalene. Symmetry 2020, 12, 1924.
- Alder, R.W.; Bowman, P.S.; Steele, W.R.; Winterman, D.R. The remarkable basicity of 1,8-bis(dimethylamino)naphthalene. Chem. Commun. 1968, 723–724.
- Pozharskii, A.F. Naphthalene “Proton Sponges”. Russ. Chem. Rev. 1998, 67, 1–27.
- Howard, S.T. Conformers, Energetics, and Basicity of 2,2′-Bipyridine. J. Am. Chem. Soc. 1996, 118, 10269–10274.
- Lesnichin, S.B.; Kamdem, N.; Mauder, D.; Denisov, G.S.; Shenderovich, I.G. Studies of Adsorption of 2,2′-Bipyridyl on the Surface of Highly Regulated Silica Matrix of the MCM-41 Type by Means of 15N NMR Spectroscopy. Russ. J. Gen. Chem. 2010, 80, 2027–2031.
- Kühn, F.E.; Groarke, M.; Bencze, E.; Herdtweck, E.; Prazeres, A.; Santos, A.M.; Calhorda, M.J.; Romao, C.C.; Goncalves, I.S.; Lopes, A.D.; et al. Octahedral Bipyridine and Bipyrimidine Dioxomolybdenum(VI) Complexes: Characterization, Application in Catalytic Epoxidation, and Density Functional Mechanistic Study. Chem. Eur. J. 2002, 8, 2370–2383.
- Lavender, E.S.; Glidewell, C.; Ferguson, G. 1,3,5-Trihydroxybenzene-2,2′-Bipyridyl (1/2): A Hydrogen-Bonded Structure Based on a Stem-and-Leaves Motif. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1998, 54, 1637–1639.
- Vogler, A.; Shenderovich, I.G. Photochemistry of deprotonated rhenium(I) (3,3’-dihydroxy-2,2’-bipyridine) tricarbonyl chloride. Photoisomerization at the chelate in basic solution. Inorg. Chim. Acta 2014, 421, 496–499.
- Morancho, R.; Pouvreau, P.; Constant, G.; Jaud, J.; Galy, J. Synthese et etude structurale d’un nouveau compose du silicium: Di-2,2′-bipyridine silicium. J. Organomet. Chem. 1979, 166, 329–338.
- Barker, D.J.; Summers, L.A.; Cooney, R.P. Conformations of 2,2′-bipyridine in acidic media. J. Mol. Struct. 1987, 159, 249–254.
- Lesnichin, S.B.; Tolstoy, P.M.; Limbach, H.-H.; Shenderovich, I.G. Counteranion-Dependent Mechanisms of Intramolecular Proton Transfer in Aprotic Solution. Phys. Chem. Chem. Phys. 2010, 12, 10373–10379.
- Chan, B.; Del Bene, J.E.; Radom, L. What factors determine whether a proton-bound homodimer has a symmetric or an asymmetric hydrogen bond? Mol. Phys. 2009, 107, 1095–1105.
- Chan, B.; Del Bene, J.E.; Radom, L. Proton-Bound Homodimers: How Are the Binding Energies Related to Proton Affinities? J. Am. Chem. Soc. 2007, 129, 12197–12199.
- Chan, B.; Del Bene, J.E.; Radom, L. Proton-bound homodimers involving second-row atoms. Theor. Chem. Acc. 2012, 131, 1088.
- Grabowski, S.J.; Ugalde, J.M. High-level ab initio calculations on low barrier hydrogen bonds and protonbound homodimers. Chem. Phys. Lett. 2010, 493, 37–44.
- Cotton, C.E.; Francisco, J.S.; Klemperer, W. Computational study of the linear proton bound ion–molecule complexes of HCNH+ with HCN and HNC. J. Chem. Phys. 2013, 139, 014304.
- Denisov, G.S.; Melikova, S.M.; Rutkovskii, K.S.; Tokhadze, K.G. Spectral Diagnostics of the Dynamics of the Formation of a Homoconjugated Complex [HCN.H.NCH]+. Opt. Spectrosc. 2020, 128, 467–469.
- Terrill, K.; Nesbitt, D.J. Ab initio anharmonic vibrational frequency predictions for linear proton-bound complexes OC–H+–CO and N2–H+–N2. Phys. Chem. Chem. Phys. 2010, 12, 8311–8322.
- Fridgen, T.D.; Parnis, J.M. Electron bombardment matrix isolation of Rg/Rg′/methanol mixtures (Rg=Ar, Kr, Xe): Fourier-transform infrared characterization of the proton-bound dimers Kr2H+, Xe2H+, (ArHKr)+ and (ArHXe)+ in Ar matrices and (KrHXe)+ and Xe2H+ in Kr matrices. J. Chem. Phys. 1998, 109, 2155.
- Kunttu, H.; Seetula, J.; Räsänen, M.; Apkarian, V.A. Photogeneration of ions via delocalized charge transfer states. I. Xe2H+ and Xe2D+ in solid Xe. J. Chem. Phys. 1992, 96, 5630.
- Grabowski, S.J. [FHF]-—The Strongest Hydrogen Bond under the Influence of External Interactions. Crystals 2016, 6, 3.
- Shenderovich, I.G.; Smirnov, S.N.; Denisov, G.S.; Gindin, V.A.; Golubev, N.S.; Dunger, A.; Reibke, R.; Kirpekar, S.; Malkina, O.L.; Limbach, H.-H. Nuclear Magnetic Resonance of Hydrogen Bonded Clusters between F- and (HF)n: Experiment and Theory. Ber. Bunsenges. Phys. Chem. Chem. Phys. 1998, 102, 422–428.
- Panich, A.M. NMR study of the F-H···F hydrogen bond. Relation between hydrogen atom position and F-H···F bond length. Chem. Phys. 1995, 196, 511–519.
- Fujiwara, F.Y.; Martin, J.S. Heterobihalide ions. Nuclear magnetic resonance spectroscopy of strong hydrogen bonds. J. Am. Chem. Soc. 1974, 96, 7625–7631.
- Limbach, H.-H.; Pietrzak, M.; Sharif, S.; Tolstoy, P.M.; Shenderovich, I.G.; Smirnov, S.N.; Golubev, N.S.; Denisov, G.S. NMR-Parameters and Geometries of OHN and ODN Hydrogen Bonds of Pyridine-Acid Complexes. Chem. Eur J. 2004, 10, 5195–5204.
- Akcakayiran, D.; Mauder, D.; Hess, C.; Sievers, T.K.; Kurth, D.G.; Shenderovich, I.; Limbach, H.-H.; Findenegg, G.H. Carboxylic Acid-Doped SBA-15 Silica as a Host for Metallo-supramolecular Coordination Polymers. J. Phys. Chem. B 2008, 112, 14637–14647.
- Ip, B.C.K.; Andreeva, D.V.; Buntkowsky, G.; Akcakayiran, D.; Findenegg, G.H.; Shenderovich, I.G. NMR Study of Proton Transfer to Strong Bases on Inner Surfaces of MCM-41. Micropor. Mesopor. Mater. 2010, 134, 22–28.
- Andreeva, D.V.; Ip, B.; Gurinov, A.A.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G.; Limbach, H.-H. Geometrical Features of Hydrogen Bonded Complexes Involving Sterically Hindered Pyridines. J. Phys. Chem. A 2006, 110, 10872–10879.
- Lorente, P.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Buntkowsky, G.; Limbach, H.-H. 1H/15N NMR Chemical Shielding, Dipolar 15N,2H Coupling and Hydrogen Bond Geometry Correlations in a Novel Serious of Hydrogen-Bonded Acid-Base Complexes of Collidine with Carboxylic Acids. Magn. Reson. Chem. 2001, 39, S18–S29.
- Lesnichin, S.B.; Shenderovich, I.G.; Muljati, T.; Limbach, H.-H.; Silverman, D. Intrinsic Proton Donating Power of Zinc-bound Water in a Carbonic Anhydrase Active Site Model Estimated by NMR. J. Am. Chem. Soc. 2011, 133, 11331–11338.
- Tolstoy, P.M.; Smirnov, S.N.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Limbach, H.-H. NMR Studies of Solid State—Solvent and H/D Isotope Effects on Hydrogen Bond Geometries of 1:1 Complexes of Collidine with Carboxylic Acids. J. Molec. Struct. 2004, 700, 19–27.
- Gurinov, A.A.; Rozhkova, Y.A.; Zukal, A.; Čejka, J.; Shenderovich, I.G. Mutable Lewis and Brønsted Acidity of Aluminated SBA-15 as Revealed by NMR of Adsorbed Pyridine-15N. Langmuir 2011, 27, 12115–12123.
- Shenderovich, I.G.; Buntkowsky, G.; Schreiber, A.; Gedat, E.; Sharif, S.; Albrecht, J.; Golubev, N.S.; Findenegg, G.H.; Limbach, H.-H. Pyridine-15N—a Mobile NMR Sensor for Surface Acidity and Surface Defects of Mesoporous Silica. J. Phys. Chem. B 2003, 107, 11924–11939.
- Chan-Huot, M.; Dos, A.; Zander, R.; Sharif, S.; Tolstoy, P.M.; Compton, S.; Fogle, E.; Toney, M.D.; Shenderovich, I.; Denisov, G.S.; et al. NMR Studies of Protonation and Hydrogen Bond States of Internal Aldimines of Pyridoxal 5′-Phosphate Acid−Base in Alanine Racemase, Aspartate Aminotransferase, and Poly-L-lysine. J. Am. Chem. Soc. 2013, 135, 18160–18175.
- Limbach, H.-H.; Chan-Huot, M.; Sharif, S.; Tolstoy, P.M.; Shenderovich, I.G.; Denisov, G.S. Critical Hydrogen Bonds and Protonation States of Pyridoxal 5′-phosphate Revealed by NMR. Biochim. Biophys. Acta 2011, 1814, 1426–1437.
- Ip, B.C.K.; Shenderovich, I.G.; Tolstoy, P.M.; Frydel, J.; Denisov, G.S.; Buntkowsky, G.; Limbach, H.-H. NMR Studies of Solid Pentachlorophenol-4-Methylpyridine Complexes Exhibiting Strong OHN Hydrogen Bonds: Geometric H/D Isotope Effects and Hydrogen Bond Coupling Cause Isotopic Polymorphism. J. Phys. Chem. A 2012, 116, 11370–11387.
- Bismarck, A.; Aranberri-Askargorta, I.; Springer, J.; Lampke, T.; Wielage, B.; Stamboulis, A.; Shenderovich, I.; Limbach, H.-H. Surface Characterization of Flax, Hemp and Cellulose Fibers; Surface Properties and the Water Uptake Behavior. Polym. Compos. 2002, 23, 872–894.
- Gurinov, A.A.; Denisov, G.S.; Borissova, A.O.; Goloveshkin, A.S.; Greindl, J.; Limbach, H.-H.; Shenderovich, I.G. NMR Study of Solvation Effect on the Geometry of Proton-Bound Homodimers of Increasing Size. J. Phys. Chem. A 2017, 121, 8697–8705.
- Attah, I.K.; Platt, S.P.; Meot-Ner, M.; El-Shall, M.S.; Aziz, S.G.; Alyoubi, A.O. Proton-bound dimers of nitrogen heterocyclic molecules: Substituent effects on the structures and binding energies of homodimers of diazine, triazine, and fluoropyridine. J. Chem. Phys. 2014, 140, 114313.
- Pollice, R.; Bot, M.; Kobylianskii, I.J.; Shenderovich, I.; Chen, P. Attenuation of London Dispersion in Dichloromethane Solutions. J. Am. Chem. Soc. 2017, 139, 13126–13140.
- Gurinov, A.A.; Lesnichin, S.B.; Limbach, H.-H.; Shenderovich, I.G. How Short is the Strongest Hydrogen Bond in the Proton-Bound Homodimers of Pyridine Derivatives? J. Phys. Chem. A 2014, 118, 10804–10812.
- Kong, S.; Borissova, A.O.; Lesnichin, S.B.; Hartl, M.; Daemen, L.L.; Eckert, J.; Antipin, M.Y.; Shenderovich, I.G. Geometry and Spectral Properties of the Protonated Homodimer of Pyridine in the Liquid and Solid States. A Combined NMR, X-ray Diffraction and Inelastic Neutron Scattering Study. J. Phys. Chem. A 2011, 115, 8041–8048.
- Shenderovich, I.G.; Limbach, H.-H.; Smirnov, S.N.; Tolstoy, P.M.; Denisov, G.S.; Golubev, N.S. H/D Isotope Effects on the Low-Temperature NMR Parameters and Hydrogen Bond Geometries of (FH)2F- and (FH)3F- Dissolved in CDF3/CDF2Cl. Phys. Chem. Chem. Phys. 2002, 4, 5488–5497.
- Perera, S.A.; Bartlett, R.J. NMR spin-spin coupling constants for hydrogen bonds of [F(HF)n]-, n=1-4, clusters. J. Am. Chem. Soc. 2000, 122, 1231–1232.
- Mulloyarova, V.V.; Ustimchuk, D.O.; Filarowski, A.; Tolstoy, P.M. H/D Isotope Effects on 1H-NMR Chemical Shifts in Cyclic Heterodimers and Heterotrimers of Phosphinic and Phosphoric Acids. Molecules 2020, 25, 1907.
- Machida, S.; Sohmiya, M.; Ide, Y.; Sugahara, Y. Solid-State 31P Nuclear Magnetic Resonance Study of Interlayer Hydroxide Surfaces of Kaolinite Probed with an Interlayer Triethylphosphine Oxide Monolayer. Langmuir 2018, 34, 12694–12701.
- Seitz, A.E.; Hippauf, F.; Kremer, W.; Kaskel, S.; Scheer, M. Facile storage and release of white phosphorus and yellow arsenic. Nat. Commun. 2018, 9, 361.
- Moussa, M.E.; Shelyganov, P.A.; Wegley, B.; Seidl, M.; Scheer, M. The Potential of the Diphosphorus Complex [Cp2W2(CO)4( η2-P2)] as an Organometallic Connecter in Supramolecular Chemistry. Eur. J. Inorg. Chem. 2019, 2019, 4241–4248.
- Guenther, J.; Reibenspies, J.; Blümel, J. Synthesis and characterization of tridentate phosphine ligands incorporating long methylene chains and ethoxysilane groups for immobilizing molecular rhodium catalysts. Mol. Catal. 2019, 479, 110629.
- Cluff, K.J.; Bhuvanesh, N.; Blümel, J. Monometallic Ni-0 and Heterobimetallic Ni-0/Au-I Complexes of Tripodal Phosphine Ligands: Characterization in Solution and in the Solid State and Catalysis. Chem. Eur. J. 2015, 21, 10138–10148.
- Pazderski, L. 15N and 31P NMR Coordination Shifts in Transition Metal Complexes with Nitrogen- and Phosphorus-Containing Heterocycles. Annu. Rep. NMR Spectrosc. 2013, 80, 33–179.
- Hubbard, P.J.; Benzie, J.W.; Bakhmutov, V.I.; Blümel, J. Disentangling Different Modes of Mobility of Triphenylphosphine Oxide Adsorbed on Alumina. J. Chem. Phys. 2020, 152, 054718.
- Kharel, S.; Cluff, K.J.; Bhuvanesh, N.; Gladysz, J.A.; Blümel, J. Structures and Dynamics of Secondary and Tertiary Alkylphosphine Oxides Adsorbed on Silica. Chem. Asian J. 2019, 14, 2704–2711.
- Hilliard, C.R.; Kharel, S.; Cluff, K.; Bhuvanesh, N.; Gladysz, J.; Bluemel, J. Structures and Unexpected Dynamic Properties of Phosphine Oxides Adsorbed on Silica Surfaces. Chem. Eur. J. 2014, 20, 17292–17295.
- Shenderovich, I.G. For Whom a Puddle Is the Sea? Adsorption of Organic Guests on Hydrated MCM-41 Silica. Langmuir 2020, 36, 11383–11392.
- Shenderovich, I.G. Effect of Noncovalent Interactions on the 31P Chemical Shift Tensor of Phosphine Oxides, Phosphinic, Phosphonic, and Phosphoric Acids, and Their Complexes with Lead(II). J. Phys. Chem. C 2013, 117, 26689–26702.
- Shenderovich, I.G. Electric field effect on 31P NMR magnetic shielding. J. Chem. Phys. 2020, 153, 184501.
- Chernyshov, I.Y.; Vener, M.V.; Shenderovich, I.G. Local-structure effects on 31P NMR chemical shift tensors in solid state. J. Chem. Phys. 2019, 150, 144706.
- Sharif, S.; Shenderovich, I.G.; González, L.; Denisov, G.S.; Silverman, D.N.; Limbach, H.-H. NMR and Ab initio Studies of Small Complexes Formed between Water and Pyridine Derivatives in Solid and Liquid Phase. J. Phys. Chem. A 2007, 111, 6084–6093.
- Grabowski, S.J. Pnicogen and tetrel bonds—tetrahedral Lewis acid centres. Struct. Chem. 2019, 30, 1141–1152.
- Grabowski, S.J. Triel bond and coordination of triel centres—Comparison with hydrogen bond interaction. Coord. Chem. Rev. 2020, 407, 213171.
- Pelties, S.; Maier, T.; Herrmann, D.; de Bruin, B.; Rebreyend, C.; Gärtner, S.; Shenderovich, I.G.; Wolf, R. Selective P4 Activation by a Highly Reduced Cobaltate: Synthesis of Dicobalt Tetraphosphido Complexes. Chem. Eur. J. 2017, 23, 6094–6102.
- Maier, T.M.; Sandl, S.; Shenderovich, I.G.; von Wangelin, A.J.; Weigand, J.J.; Wolf, R. Amine-Borane Dehydrogenation and Transfer Hydrogenation Catalyzed by alpha-Diimine Cobaltates. Chem. Eur. J. 2019, 25, 238–245.
- Medvedev, A.G.; Churakov, A.V.; Prikhodchenko, P.V.; Lev, O.; Vener, M.V. Crystalline Peroxosolvates: Nature of the Coformer, Hydrogen-Bonded Networks and Clusters, Intermolecular Interactions. Molecules 2021, 26, 26.
- Giba, I.S.; Tolstoy, P.M. Self-Assembly of Hydrogen-Bonded Cage Tetramers of Phosphonic Acid. Symmetry 2021, 13, 258.
- Kizior, B.; Panek, J.J.; Szyja, B.M.; Jezierska, A. Structure-Property Relationship in Selected Naphtho- and Anthra-Quinone Derivatives on the Basis of Density Functional Theory and Car–Parrinello Molecular Dynamics. Symmetry 2021, 13, 564.
- Alkorta, I.; Elguero, J.; Frontera, A. Not Only Hydrogen Bonds: Other Noncovalent Interactions. Crystals 2020, 10, 180.