The mammalian target of the rapamycin (mTOR) system regulates various cellular functions, such as growth, proliferation, metabolism and survival/death. In systemic organs, it is critically involved in multiple processes, including neurogenesis, nutrition and immunity. In the brain, its roles are essential in the cerebral cortical development, synaptic functions, and brain activities such as learning, cognition and social functions.
- mTORopathy
- mTOR inhibitor
- TSC
- epilepsy
- intellectual disability
- autism
- epileptic encephalopathy
1. Introduction
2. Pathology and Clinical Picture of TSC
From a pathologic point of view, TSC is characterized by the multifocal occurrence of benign tumors (hamartomas) and focal dysplastic lesions (hamartias) in various organs, such as the skin, brain, eye, heart, lungs, and kidneys [29][1]. In the vast majority of organs, these morphologic lesions, especially tumors, are the sole cause of functional problems, such as dysmorphism, rupture, and pressure to the surrounding normal tissues. In this context, the cerebrum is a remarkable exception as many patients with TSC have brain dysfunction, such as ID and ASD [30[2][3],31], without an apparent causal relationship with anatomical lesions. Thus, the clinical findings of TSC can be classified into three groups: (1) hamartoma, (2) focal dysplasia, and (3) brain dysfunction.
With regard to the occurrence/distribution of lesions and the severity of brain dysfunction, these clinical symptoms are remarkably variable among patients, which is also true for familial cases with the same mutation. The genotype–phenotype correlation is reported to be small [32[4][5][6],33,34], and the reason for the inter-case variability remains largely unknown. Each lesion or symptom shows a time course that is clearly age dependent. For instance, cardiac rhabdomyoma arises in the fetal period, facial angiofibroma in childhood, and pulmonary lymphangioleiomyomatosis (LAM) in adulthood
Many (about 80%) TSC patients have epileptic seizures of variable types. Typical clinical pictures are West syndrome (infantile spasms) in infancy, and focal epilepsy, which may occur in any age group [30,31][2][3]. In the vast majority of patients, the epileptic focus is located in or adjacent to a cortical tuber (focal dysplasia of the cerebral cortex). Epilepsy is often resistant to antiepileptic drugs, requiring neurosurgical treatment in many cases.
The level of intelligence is variable among patients, ranging from normal to profound ID. ID is present in more than half of patients, and ASD in about half. Even patients with normal intelligence have a variety of behavioral, cognitive, and psychosocial problems, which are collectively called TSC-associated neuropsychiatric disorders (TAND) [35][7].
Approximately 10% of patients have subependymal giant cell astrocytoma (SEGA), a benign tumor on the wall of the lateral ventricle. A large SEGA may cause hydrocephalus and clinical signs of increased intracranial pressure. For the treatment of SEGA, the first choice is surgical resection of the tumor. Chemotherapy with an mTOR inhibitor, everolimus, is also efficacious, and has recently become another choice of therapy [25][8].
3. Etiology and Pathogenesis of TSC
TSC is caused by various loss-of-function mutations in the two genes,TSC1andTSC2[33,34][5][6]. A genotype–phenotype relationship has been noted, but it is small. There is no qualitative difference betweenTSC1andTSC2. However,TSC2mutations tend to show more severe brain symptoms and a larger propensity to develop tumors thanTSC1mutations [32,33,34][4][5][6].
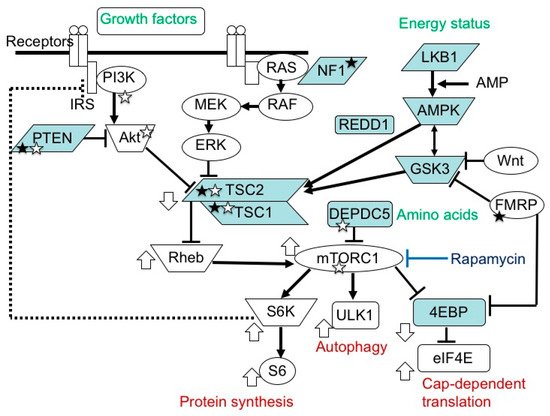
TSC1andTSC2encode for tumor suppressors, hamartin and tuberin, respectively. These proteins bind to form a complex and then stabilize each other [2,3][9][10]. TSC-associated tumors show a decreased expression in both [36,37][11][12]. The TSC1/TSC2 complex is located in the midstream of the mTOR pathway and negatively regulates the activity of the system [2,3,4,5,6][9][10][13][14][15] (Figure 1).
In TSC, a decrease in the regulatory function of TSC1/TSC2 causes the chronic hyperactivation of the downstream mTOR system, which affects cellular proliferation [38][16], migration [39[17][18],40], glucose uptake/metabolism [41][19], and angiogenesis [42][20], leading to tumorigenesis and dysgenesis.
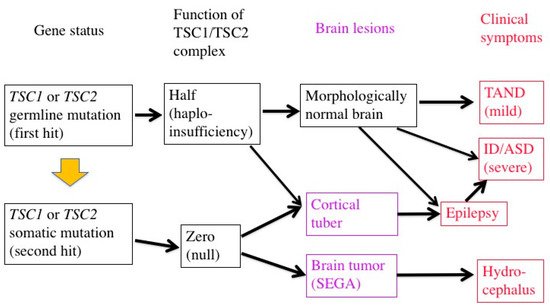
In patients with TSC, all the somatic cells have a germline mutation (first hit) in one allele of either theTSC1orTSC2gene, which can cause haploinsufficiency of the TSC1/TSC2 complex. When an additional somatic mutation (second hit) occurs during mitosis, the function of the TSC1/TSC2 complex becomes null.
Previous studies have demonstrated that TSC-associated tumors (hamartomas) occur according to the two-hit hypothesis [22,23,43][21][22][23]. The second hit is typically a small deletion of either 9q34 (TSC1) or 16p13.3 (TSC2), causing loss of heterozygosity (LOH), the incidence of which is reportedly high in kidney tumors (renal angiomyolipoma) but low in brain tumors (SEGA).
In cerebral dysplastic lesions, namely cortical tubers, LOH has not been found [22,23,43,44][21][22][23][24]. Studies using laser capture microdissection have found point mutations (but not LOH) in abnormal giant cells (astrocyte-like balloon cells and cytomegalic neurons), which are a histopathological hallmark of cortical tubers [24,45][25][26]. A cortical tuber comprises a small number of abnormal giant cells with null TSC1/TSC2 function, and a large number of normal-sized neurons/glial cells with haploinsufficiency [46][27]. The interaction of both cell types may account for the epileptogenicity of cortical tubers (Figure 2).
mTOR inhibitors are useful in the treatment of TSC-associated tumors in the brain, heart, and kidneys (everolimus) [25,26[8][28][29],47], as well as in the skin and lungs (sirolimus) [48,49][30][31]. Furthermore, clinical studies have recently shown that mTOR inhibitors are effective not only for tumors, phenotypes predominantly resulting from the second hit, but also for brain dysfunction (epilepsy) caused by a combination of the first and second hits [27][32]. The molecular basis of the efficacy is that the main stream of the mTOR system is essentially single between the TSC1/TSC2 complex and mTORC1 (Figure 1).
4. Brain Dysfunction in TSC
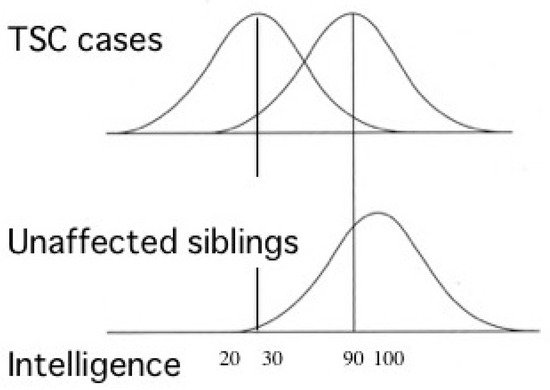
Most TSC patients have a variety of behavioral, cognitive, and/or psychiatric problems, collectively termed as TAND. ID and ASD are the most common, affecting about 80% and 40% of patients, respectively. Notably, the distribution of intelligence quotient (IQ) is bimodal, dividing patients into two groups: profound ID with IQ (less than 30), and normal/subnormal mentality with a slight reduction in average IQ (around 90) [50] (Figure 3). Epidemiologic studies have shown that early-onset epilepsy (represented by West syndrome) is much more common in the former group than in the latter [51,52].
TSC is a common cause of symptomatic ASD, second only to fragile X syndrome in prevalence. The sex ratio (male:female) of entire ASD is 4:1, whereas that of TSC-associated ASD is 1:1 [53,54][33][34]. In TSC, ASD is more common in patients with early-onset epilepsy [35,55][7][35].
As described above, a cortical tuber consists of both abnormal giant cells caused by a somatic mutation (second hit) and normal-sized neurons/glial cells with a germline mutation (first hit) only. Pathologic collaboration (networking) between severely dysfunctional, abnormal giant cells and mildly dysfunctional, normal-sized neurons/glial cells forms the epileptogenic focus. Thus, in the brain, mild dysfunction due to germline mutations (first hit) can cause clinical symptoms, in sharp contrast to other organs where all the symptoms are due to somatic mutations (second hit). There is evidence for the dysfunction of autophagy/cell clearing, which is implicated in epileptogenesis [12,13,58,59][36][37][38][39].
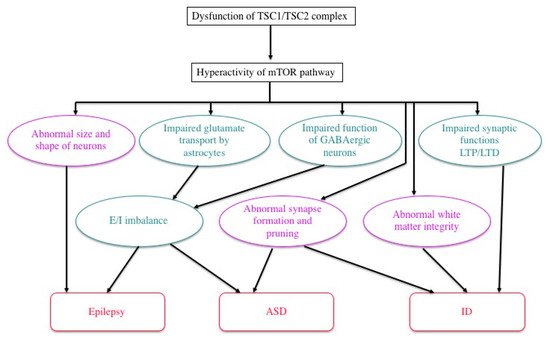
Previous studies in human patients and animal models, either in vivo or in vitro, have documented various abnormalities of TSC neurons and/or glial cells (Figure 4). The expression and function of gamma aminobutyric acid (GABA) receptors are abnormal in abnormal giant cells of cortical tubers [61,62,63,64,65][40][41][42][43][44]. Synapse formation is defective in human cortical tubers [66][45], as well as in cultured neurons ofTsc1andTsc2knockout mice [67]. White matter also shows abnormal findings, which are implicated in the pathogenesis of ASD.
To date, numerous animal models of TSC have been developed and used. One model, Eker rat, occurred spontaneously, whereas all the other models were produced artificially by genetic engineering. For translational research on the treatment of TSC-associated brain functions, we and other investigators often used mice produced by conventional gene knockout, due to their excellent construct validity.
The Eker rat, harboring a germlineTsc2mutation, was originally found as a model of the hereditary cancer, renal cell carcinoma [74,75,76][46][47][48]. Compared to human TSC, the brain phenotype of the Eker rat is much milder, with a rare occurrence of cortical tuber-like lesions (focal cortical dysplasia) [77][49], an absence of epileptic seizures occurring spontaneously, and only a mild deficit in social interaction. Experimental studies introducing a second hit stimulus to the developing brain revealed that irradiation, but not a carcinogen, can increase the incidence of abnormal giant cells [78,79][50][51]. When the developing brain was pharmacologically exposed to severe epilepsy, the rat displayed ASD-like social deficit behavior [80][52].
Except for severalTsc1+/−mouse models [81[53][54],82], all heterozygous knockout mice for eitherTsc1orTsc2mutants showed neither cortical tubers nor spontaneous epileptic seizures. Thus, mostTsc1+/−andTsc2+/−mice are not suitable as models of TSC-associated epilepsy, or brain symptoms caused by a combination of the first hit (normal-sized neurons and glial cells) and the second hit (abnormal giant cells). For example, aTsc2+/−mouse showed deficits in some hippocampal functions, such as spatial learning and contextual discrimination. They also establishedTsc1+/−andTsc2+/−mice as models of mild TAND in the absence of early-onset epilepsy and illustrated their value in translational research of therapies for TAND.
Since the beginning of this century, pharmacological treatment with mTOR inhibitors has rapidly progressed, especially for TSC-associated hamartomas: everolimus for brain tumors (SEGA) [25][8] and renal tumors (AML) [26][28], and sirolimus (rapamycin) for skin tumors (facial angiofibroma, topical) [48][30] and pulmonary tumors (LAM) [49][31]. When treated with mTOR inhibitors, these tumors shrink over several weeks or months. At worst, the tumor size remains unchanged [25,26][8][28], which nevertheless is usually acceptable in clinical practice because the tumors are benign in nature.
During clinical trials of oral (systemic) administration mTOR inhibitors, researchers noted their ancillary efficacy on brain dysfunctions, such as a decrease in epileptic seizures and the improvement of ASD symptoms. A phase 3, international clinical trial on the efficacy of everolimus for focal epilepsy (named EXIST-3) was conducted, and successfully demonstrated the usefulness of everolimus in the treatment of TSC-associated epilepsy by showing a clinically meaningful reduction in seizure frequency [27][32]. On the other hand, the inter-case variability was even larger in epilepsy than in cerebral and renal tumors; epileptic seizures totally disappeared in the best case, whereas they increased by three times in the worst case. With regard to both epilepsy and ASD, the reasons for the large inter-case variability remain unclear.
References
- Gomez, M.R. Definition and criteria for diagnosis. In Tuberous Sclerosis Complex, 3rd ed.; Gomez, M.R., Sampson, J.R., Whittemore, V.H., Eds.; Oxford University Press: New York, NY, USA, 1999; pp. 10–23.
- Gomez, M.R. Natural history of cerebral tuberous sclerosis. In Tuberous Sclerosis Complex, 3rd ed.; Gomez, M.R., Sampson, J.R., Whittemore, V.H., Eds.; Oxford University Press: New York, NY, USA, 1999; pp. 29–46.
- Wataya-Kaneda, M.; Tanaka, M.; Hamasaki, T.; Katayama, I. Trends in the prevalence of tuberous sclerosis complex manifestations: An epidemiological study of 166 Japanese patients. PLoS ONE 2013, 8.
- Jones, A.C.; Shyamsundar, M.M.; Thomas, M.W.; Maynard, J.; Idziaszczyk, S.; Tomkins, S.; Sampson, J.R.; Cheadle, J.P. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 1999, 64, 1305–1315.
- Dabora, S.L.; Jozwiak, S.; Franz, D.N.; Roberts, P.S.; Nieto, A.; Chung, J.; Choy, Y.S.; Reeve, M.P.; Thiele, E.; Egelhoff, J.C.; et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am. J. Hum. Genet. 2001, 68, 64–80.
- Niida, Y.; Wakisaka, A.; Tsuji, T.; Yamada, H.; Kuroda, M.; Mitani, Y.; Okumura, A.; Yokoi, A. Mutational analysis of TSC1 and TSC2 in Japanese patients with tuberous sclerosis complex revealed higher incidence of TSC1 patients than previously reported. J. Hum. Genet. 2013, 58, 216–225.
- Curatolo, P. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015, 14, 733–745.
- Franz, D.N.; Belousova, E.; Sparagana, S.; Bebin, E.M.; Frost, M.; Kuperman, R.; Witt, O.; Kohrman, M.H.; Flamini, J.R.; Wu, J.Y.; et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): A multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2013, 381, 125–132.
- van Slegtenhorst, M.; Nellist, M.; Nagelkerken, B.; Cheadle, J.; Snell, R.; van den Ouweland, A.; Reuser, A.; Sampson, J.; Halley, D.; van der Sluijs, P. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum. Mol. Genet. 1998, 7, 1053–1057.
- Wienecke, R.; König, A.; DeClue, J.E. Identification of tuberin, the tuberous sclerosis-2 product. Tuberin possesses specific Rap1GAP activity. J. Biol. Chem. 1995, 270, 16409–16414.
- Mizuguchi, M.; Kato, M.; Yamanouchi, H.; Ikeda, K.; Takashima, S. Tuberin immunohistochemistry in brain, kidneys and heart with or without tuberous sclerosis. Acta Neuropathol. 1997, 94, 525–531.
- Mizuguchi, M.; Ikeda, K.; Takashima, S. Simultaneous loss of hamartin and tuberin from the cerebrum, kidney and heart with tuberous sclerosis. Acta Neuropathol. 2000, 99, 503–510.
- Xiao, G.H.; Shoarinejad, F.; Jin, F.; Golemis, E.A.; Yeung, R.S. The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. J. Biol. Chem. 1997, 272, 6097–6100.
- Benvenuto, G.; Li, S.; Brown, S.J.; Braverman, R.; Vass, W.C.; Cheadle, J.P.; Halley, D.J.; Sampson, J.R.; Wienecke, R.; DeClue, J.E. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene 2000, 19, 6306–6316.
- Tee, A.R.; Fingar, D.C.; Manning, B.D.; Kwiatkowski, D.J.; Cantley, L.C.; Blenis, J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13571–13576.
- Hengstschläger, M.; Rodman, D.M.; Miloloza, A.; Hengstschläger-Ottnad, E.; Rosner, M.; Kubista, M. Tuberous sclerosis gene products in proliferation control. Mutat. Res. 2001, 488, 233–239.
- Mizuguchi, M.; Yamanouchi, H.; Becker, L.E.; Itoh, M.; Takashima, S. Doublecortin immunoreactivity in giant cells of tuberous sclerosis and focal cortical dysplasia. Acta Neuropathol. 2002, 104, 418–424.
- Ohsawa, M.; Kobayashi, T.; Okura, H.; Igarashi, T.; Mizuguchi, M.; Hino, O. TSC1 controls distribution of actin fibers through its effect on function of Rho family of small GTPases and regulates cell migration and polarity. PLoS ONE 2013, 8.
- Jiang, X.; Kenerson, H.; Aicher, L.; Miyaoka, R.; Eary, J.; Bissler, J.; Yeung, R.S. The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am. J. Pathol. 2008, 172, 1748–1756.
- Brugarolas, J.; Kaelin, W.G., Jr. Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell 2004, 6, 7–10.
- Green, A.J.; Smith, M.; Yates, J.R. Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat. Genet. 1994, 6, 193–196.
- Henske, E.P.; Scheithauer, B.W.; Short, M.P.; Wollmann, R.; Nahmias, J.; Hornigold, N.; Slegtenhorst, M.; Welsh, C.T.; Kwiatkowski, D.J. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am. J. Hum. Genet. 1996, 59, 400–406.
- Niida, Y.; Stemmer-Rachamimov, A.O.; Logrip, M.; Tapon, D.; Perez, R.; Kwiatkowski, D.J.; Sims, K.; MacCollin, M.; Louis, D.N.; Ramesh, V. Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am. J. Hum. Genet. 2001, 69, 493–503.
- Wolf, H.K.; Normann, S.; Green, A.J.; von Bakel, I.; Blümcke, I.; Pietsch, T.; Wiestler, O.D.; von Deimling, A. Tuberous sclerosis-like lesions in epileptogenic human neocortex lack allelic loss at the TSC1 and TSC2 regions. Acta Neuropathol. 1997, 93, 93–96.
- Crino, P.B.; Aronica, E.; Baltuch, G.; Nathanson, K.L. Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 2010, 74, 1716–1723.
- Mizuguchi, M.; Mori, M.; Nozaki, Y.; Momoi, M.Y.; Itoh, M.; Takashima, S.; Hino, O. Absence of allelic loss in cytomegalic neurons of cortical tuber in the Eker rat model of tuberous sclerosis. Acta Neuropathol. 2004, 107, 47–52.
- Mizuguchi, M.; Takashima, S. Neuropathology of tuberous sclerosis. Brain Dev. 2001, 23, 508–515.
- Bissler, J.J.; Kingswood, J.C.; Radzikowska, E.; Zonnenberg, B.A.; Frost, M.; Belousova, E.; Sauter, M.; Nonomura, N.; Brakemeier, S.; de Vries, P.J.; et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): A multicentre, randomised, double-blind, placebo-controlled trial. Lancet 2013, 381, 817–824.
- Aw, F.; Goyer, I.; Raboisson, M.J.; Boutin, C.; Major, P.; Dahdah, N. Accelerated cardiac rhabdomyoma regression with everolimus in infants with tuberous sclerosis complex. Pediatr. Cardiol. 2017, 38, 394–400.
- Wataya-Kaneda, M.; Ohno, Y.; Fujita, Y.; Yokozeki, H.; Niizeki, H.; Ogai, M.; Fukai, K.; Nagai, H.; Yoshida, Y.; Hamada, I.; et al. Sirolimus gel treatment vs. placebo for facial angiofibromas in patients with tuberous sclerosis complex: A randomized clinical trial. JAMA Dermatol. 2018, 154, 781–788.
- Bissler, J.J.; McCormack, F.X.; Young, L.R.; Elwing, J.M.; Chuck, G.; Leonard, J.M.; Schmithorst, V.J.; Laor, T.; Brody, A.S.; Bean, J.; et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med. 2008, 358, 140–151.
- French, J.A.; Lawson, J.A.; Yapici, Z.; Ikeda, H.; Polster, T.; Nabbout, R.; Curatolo, P.; de Vries, P.J.; Dlugos, D.J.; Berkowitz, N.; et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): A phase 3, randomised, double-blind, placebo-controlled study. Lancet 2016, 388, 2153–2163.
- Lewis, J.C.; Thomas, H.V.; Murphy, K.C.; Sampson, J.R. Genotype and psychological phenotype in tuberous sclerosis. J. Med. Genet. 2004, 41, 203–207.
- de Vries, P.J.; Hunt, A.; Bolton, P.F. The psychopathologies of children and adolescents with tuberous sclerosis complex (TSC): A postal survey of UK families. Eur. Child Adolesc. Psychiatry 2007, 16, 16–24.
- de Vries, P.J.; Whittemore, V.H.; Leclezio, L.; Byars, A.W.; Dunn, D.; Ess, K.C.; Hook, D.; King, B.H.; Sahin, M.; Jansen, A. Tuberous sclerosis associated neuropsychiatric disorders (TAND) and the TAND Checklist. Pediatr. Neurol. 2015, 52, 25–35.
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Fabrizi, C.; Frati, A.; Fornai, F. mTOR-related cell-clearing systems in epileptic seizures, an update. Int. J. Mol. Sci. 2020, 21, 1642.
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141.
- McMahon, J.; Huang, X.; Yang, J.; Komatsu, M.; Yue, Z.; Qian, J.; Zhu, X.; Huang, Y. Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J. Neurosci. 2012, 32, 15704–15714.
- Yasin, S.A.; Ali, A.M.; Tata, M.; Picker, S.R.; Anderson, G.W.; Latimer-Bowman, E.; Nicholson, S.L.; Harkness, W.; Cross, J.H.; Paine, S.M.; et al. mTOR-dependent abnormalities in autophagy characterize human malformations of cortical development: Evidence from focal cortical dysplasia and tuberous sclerosis. Acta Neuropathol. 2013, 126, 207–218.
- White, R.; Hua, Y.; Scheithauer, B.; Lynch, D.R.; Henske, E.P.; Crino, P.B. Selective alterations in glutamate and GABA receptor subunit mRNA expression in dysplastic neurons and giant cells of cortical tubers. Ann. Neurol. 2001, 49, 67–78.
- Talos, D.M.; Sun, H.; Kosaras, B.; Joseph, A.; Folkerth, R.D.; Poduri, A.; Madsen, J.R.; Black, P.M.; Jensen, F.E. Altered inhibition in tuberous sclerosis and type IIb cortical dysplasia. Ann. Neurol. 2012, 71, 539–551.
- Valencia, I.; Legido, A.; Yelin, K.; Khurana, D.; Kothare, S.V.; Katsetos, C.D. Anomalous inhibitory circuits in cortical tubers of human tuberous sclerosis complex associated with refractory epilepsy: Aberrant expression of parvalbumin and calbindin-D28k in dysplastic cortex. J. Child Neurol. 2006, 21, 1058–1063.
- Wang, Y.; Greenwood, J.S.; Calcagnotto, M.E.; Kirsch, H.E.; Marmaro, N.M.; Baraban, S.C. Neocortical hyperexcitability in a human case of tuberous sclerosis complex and mice lacking neuronal expression of TSC1. Ann. Neurol. 2007, 61, 139–152.
- Zhao, J.-P.; Yoshii, A. Hyperexcitability of the local cortical circuit in mouse models of tuberous sclerosis complex. Mol. Brain 2019, 12, 6.
- Lippa, C.F.; Pearson, D.; Smith, T.W. Cortical tubers demonstrate reduced immunoreactivity for synapsin I. Acta Neuropathol. 1993, 85, 449–451.
- Hino, O.; Klein-Szanto, A.J.; Freed, J.J.; Testa, J.R.; Brown, D.Q.; Vilensky, M.; Yeung, R.S.; Tartof, K.D.; Knudson, A.G. Spontaneous and radiation-induced renal tumors in the Eker rat model of dominantly inherited cancer. Proc. Natl. Acad. Sci. USA 1993, 90, 327–331.
- Everitt, J.I.; Goldsworthy, T.L.; Wolf, D.C.; Walker, C.L. Hereditary renal cell carcinoma in the Eker rat: A rodent familial cancer syndrome. J. Urol. 1992, 148, 1932–1936.
- Kobayashi, T.; Hirayama, Y.; Kobayashi, E.; Kubo, Y.; Hino, O. A germline insertion in the tuberous sclerosis (Tsc2) gene gives rise to the Eker rat model of dominantly inherited cancer. Nat. Genet. 1995, 9, 70–74.
- Mizuguchi, M.; Takashima, S.; Yamanouchi, H.; Nakazato, Y.; Mitani, H.; Hino, O. Novel cerebral lesions in the Eker rat model of tuberous sclerosis: Cortical tuber and anaplastic gangliogioma. J. Neuropathol. Exp. Neurol. 2000, 59, 188–196.
- Wenzel, H.J.; Patel, L.S.; Robbins, C.A.; Emmi, A.; Yeung, R.S.; Schwartzkroin, P.A. Morphology of cerebral lesions in the Eker rat model of tuberous sclerosis. Acta Neuropathol. 2004, 108, 97–108.
- Takahashi, D.J.; Dinday, M.T.; Barbaro, N.M.; Baraban, S.C. Abnormal cortical cells and astrocytomas in the Eker rat model of tuberous sclerosis complex. Epilepsia 2004, 45, 1525–1530.
- Schneider, M.; DeVries, P.J.; Schönig, K.; Rößner, V.; Waltereit, R. mTOR inhibitor reverses autistic-like social deficit behaviours in adult rats with both Tsc2 haploinsufficiency and developmental status epilepticus. Eur. Arch. Psychiatry Clin. Neurosci. 2017, 267, 455–463.
- Feliciano, D.M.; Su, T.; Lopez, J.C.; Bordey, A. Single-cell Tsc1 knockout during corticogenesis generates tuber-like lesions and reduces seizure threshold in mice. J. Clin. Investig. 2011, 121, 1596–1607.
- Gataullina, S.; Lemaire, E.; Wendling, F.; Kaminska, A.; Watrin, F.; Riquet, A.; Ville, D.; Moutard, M.L.; de Saint Martin, A.; Napuri, S.; et al. Epilepsy in young Tsc1(+/−) mice exhibits age-dependent expression that mimics that of human tuberous sclerosis complex. Epilepsia 2016, 57, 648–659.