Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 1 by Kostis Gyftopoulos.
Epithelial–mesenchymal transition (EMT) is a dynamic, reversible cellular process which is essential in normal embryonic morphogenesis and wound healing. However this multifaceted process is also implicated in cancer progression and metastasis in many tumors, including prostate cancer. Moreover, recent evidence suggests that EMT-induced cell plasticity is involved in the development of therapy resistance in prostate cancer cells.
- prostate cancer
- EMT
- metastasis
- cell plasticity
- therapy resistance
1. Introduction
Prostate cancer (PCa) is the second most frequently diagnosed male malignancy and the second leading cause of cancer mortality in men [1]. As a general rule, both incidence and mortality of PCa correlate with advancing age, with the average age at the time of diagnosis being 66 years [2]. For African American men, the incidence rates are higher when compared to White men; alarmingly, they also have the highest chance of being diagnosed at a younger age (<40 years) [3]. However, this is also true for men generally; the incidence of PCa in young men is rising, and their younger age poses a higher risk for metastatic disease and eventually a higher mortality [3]. It is estimated that mortality will double from 2018 to 2040, reaching 379,005 deaths worldwide. The highest mortality rates are expected in Africa (+124.4%) and Asia (116.7%), while the lowest incidence is expected to be registered in Europe (+58.3%) [4]. It is well known that PCa is a multifocal disease with a heterogeneous cell population. This excessive heterogeneity and plasticity which characterize prostate tumors underline the regulatory phenotypic changes in individual cells that contribute to metastasis and therapeutic resistance [5]. The critical problem in the available pharmaceutical therapeutic approach of PCa is that PCa unavoidably develops resistance to androgen deprivation therapy (ADT) at some stage and progresses to castration-resistant PCa (CRPC), which is characterized by invasiveness and metastatic spread of CRPC cells; this phenomenon still represents the major cause of PCa-related death. Thus, understanding the cellular and molecular mechanisms underlying the process of metastatic dissemination of PCa remains a challenge for better management of PCa.
Recently, ample evidence of epithelial–mesenchymal plasticity playing a role in both PCa metastatic progression and treatment resistance has been provided in several reports. Epithelial–mesenchymal transition (EMT) is a molecular cellular program essential for development, and the relevant processes are reactivated during wound healing, fibrosis, and cancer progression [6,7][6][7]. During EMT, epithelial cells lose their junctions and apical–basal polarity, restructure their cytoskeleton, undergo changes in the signaling programs that specify cell shape, and reprogram gene expression; this enhances the motility of individual cells and empowers the progression of a more invasive phenotype [8,9][8][9]. EMT is identified by a loss of epithelial markers such as cytokeratins and E-cadherin, followed by a concurrent increase in mesenchymal markers such as N-cadherin and vimentin [10]. In cancer progression, this is connected with poor clinical outcome and therapy resistance in several cancer types [11]. In PCa, for instance, loss of E-cadherin expression and overexpression of N-cadherin correlate with tumor grade and recurrence after surgery, providing a clinically significant link of EMT to aggressive clinical behavior in advanced disease [12]. The cellular processes of EMT are coordinated by several key transcription factors (e.g., TWIST, SNAI1, SNAI2, ZEB 1/2) that act in unison with several epigenetic mechanisms and post-translational protein modifications to arrange the cellular alterations [13]. The main molecular events are depicted in Figure 1.
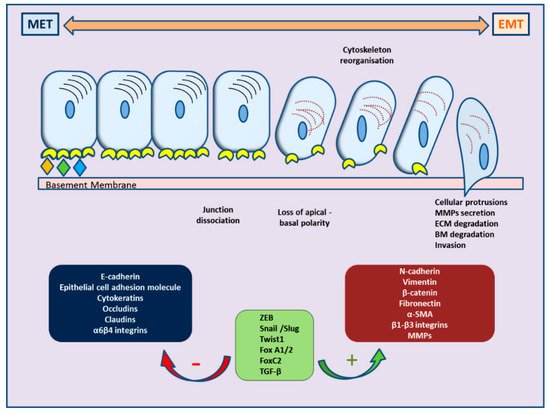
Figure 1. Schematic representation of cellular changes and main molecular events during epithelial–mesenchymal transition (EMT) and the reverse MET.
Different critical signaling pathways including TGF-β, WNT, NOTCH, and growth factors are involved in inducing EMT under specific physiological conditions. Extensive research and available data are suggesting that EMT facilitates tumor progression and metastasis in several cancer types. In this review, we summarize and refer to the main molecular events that control the EMT phenomenon in PCa, in order to better describe its role and mechanisms in PCa progression and therapeutic resistance.
2. EMT and Therapy Resistance in Prostate Cancer
EMT is evidently correlated not only with tumor metastasis but, most importantly, also with therapy resistance. The role of EMT is distinct in several tumors, including solid tumors and endocrine-related tumors such as breast and prostate carcinoma [232,233,234,235][14][15][16][17]. The stem-cell features acquired during the EMT process have revealed that the EMT program is a critical regulator of the cancer stem cell (CSC) phenotype, mainly through epigenetic changes (Figure 32). CSCs are more resistant than non-CSCs to various types of conventional therapies and this may account for treatment failure and CSC-mediated clinical relapse [236][18]. In preclinical and clinical models, chemotherapy and/or radiotherapy have been shown to eliminate the bulk population of mainly non-CSCs, while leaving behind considerable numbers of CSCs in multiple cancer types [236][18].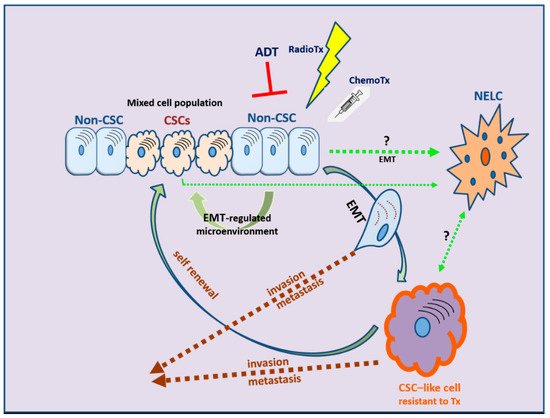
Figure 32. The development of therapy resistance: the complex interlinks among therapeutic manipulations, EMT, and cancer stem cell (CSC) development. NELC: neuro-endocrine-like cell; ADT: androgen deprivation therapy; RadioTx: radiotherapy; ChemoTx: chemotherapy.
2.1. Androgen Deprivation Therapy
The mainstay of advanced or metastatic PCa therapy remains androgen blockade, i.e., ADT. As discussed earlier, the androgen receptor plays a disputable role in the course of the disease, eventually acting as an undesired promoter of EMT, leading to CRPC status. Although solid evidence is still missing, the rationale of combination therapies that aim at both androgen and EMT pathways is intriguing. Hence, dual approaches include the use of TGF-β inhibitors that may suppress both TWIST1 and AR expression or TGF-β receptor inhibitors that exert FOXA1-like suppressor effects on TGF-β (galunisertib), the latter being currently used in a clinical trial in combination with the antiandrogen enzalutamide [49,237,238][19][20][21]. Another approach is focusing on the presence of androgen receptor splice variants or truncated receptors that are often present in CPRC. These aberrant receptors may be inhibited by novel retinamides [239,240][22][23]. In addition to retinoids, the use of novel AR antagonists that target both wild-type and mutated ligand-binding domain variants to inhibit AR nuclear translocation has led to favorable results both in vitro and in vivo [241][24]. Last but not least, the neuroendocrine differentiation (NED) eventually induced by androgen ablation is an important factor for treatment failure, with the EMT process again playing a central role. MicroRNAs, such as miR-652, are implicated in this process [242][25]. Lately, small-molecule drugs and miRNA mimics have emerged as practical tool in the regulation of miRNAs and in potentially suppressing drug resistance in PCa [243][26]. An alternative approach would be to target the Wnt/β-catenin signaling pathway associated with NE differentiation [71][27].
The EMT process in PCa has also been associated with immune evasion due to upregulation of indoleamine 2,3-expression (IDO1) accompanied by an increased number of immunosuppressive regulatory T cells [244][28]. Similar findings in bladder cancer, where IDO1 expression can upregulate ZEB2 expression probably through miR-200c signaling, provide a promising novel target against immunosuppression in PCa and other tumor types [245][29].
2.2. Chemotherapy
EMT in PCa is associated with resistance to chemotherapy, orchestrated mainly by the action of transcription factors such as ZEB1 and ZEB2, as well as by downregulation of “pro-epithelial” miRNAs, such as miR-143, miR-145, miR-29b, miR-34b, and the miR-200 family [235,246][17][30]. Loss of expression of miR-200 and miR-205c occurs during prolonged treatment with docetaxel, suggesting that their restoration may reinstate chemosensitivity by inhibiting EMT [247][31]. A different approach has been very recently supported by in vitro results using alternative compounds, such as green tea, flavonoids, and minerals (zinc). Green tea (GT) and quercetin (Q), a flavonoid from apples and onions, enhances the efficacy of docetaxel in androgen-independent PCa cells through multiple mechanisms, including the downregulation of chemoresistance-related proteins [248][32]. Quercetin was also found to overcome docetaxel resistance in PCa cells via androgen receptor and PI3K/Akt signaling pathways, reversing mesenchymal and stem-like cell changes in phenotype and reducing multidrug resistance (MDR) transporters, such as P-glycoprotein (Pgp) expression [249][33]. Moreover, quercetin was shown to suppress the EMT process, deactivating the PI3K/Akt signaling pathway and downregulating MALAT1, a long noncoding RNA [250][34]. Similarly, zinc’s synergistic effect to paclitaxel was demonstrated in PCa cell lines via EMT inhibition by downregulating the expression of TWIST1 [251][35].
2.3. Radiation Therapy
Radiotherapy is an effective treatment option for localized PCa, with favorable response rates; however, cancer cells eventually acquire radioresistance (RR) during fractionated irradiation (IR) [252][36]. Beyond its established role in tumor invasion, metastasis, and recurrence, EMT is also linked to radioresistance. EMT is closely associated with CRPC, and PCa cells with more mesenchymal markers such as Snail, Vimentin, SOX2, and N-cadherin exhibit radioresistance [253][37]. The common alterations in PCa cells comprise an increased number of cancer stem cells (CSCs), neuroendocrine differentiation (NED), and epithelial–mesenchymal transition, all of which are interlinked, as discussed earlier. Earlier studies have shown that IR itself may induce EMT in prostate tumors via activation of the cAMP response element-binding protein and cytoplasmic sequestration of the activating transcription factor 2 [254][38]. Recently, it has be suggested that fractionated irradiation causes an increased expression of pluripotency-associated genes, further inducing CSCs and driving progression of NED, which triggers RR acquisition in PCa [252][36].
The IR-induced EMT process is mediated by several transcription factors (Snail/Slug, STAT3, Twist, ZEB1, and ZEB2) that are activated by a variety of signaling pathways (i.e., Hedgehog, Notch, TGF-β, Wnt, and others) [255][39]. Recently, increased EZH2 expression in PCa was associated with metastatic recurrence following external beam radiotherapy [256][40]. The final mechanisms or radioresistance include prolongation of the production of mitochondrial ROS, which delay cell growth as a mechanism for cell death resistance, modulation of the rate of H2O2 production and the balance between O2•− and H2O2, and activation of Homeobox B9 for enhancement of DNA damage and repair responses. It has been proposed that radioresistant PCa cells adapt to higher oxidative stress via upregulation of endogenous ROS-generating enzymes and antioxidant proteins [255][39]. Recently, structural maintenance of chromosome-1 (SMC1A), a subunit of the cohesin complex involved in chromosome cohesion during cell-cycle and DNA repair, was identified as a key factor in acquired radioresistance of metastatic PCa cells. Interestingly, inhibition of SMC1A in a model of DU145 and PC3 cells limited the clonogenic, EMT, and cancer stem-like cell (CSC) properties of cancer cells and rendered them more susceptible to RT [257][41]. In a similar manner, knockdown of LOXL2, a member of the lysyl oxidase gene family, enhanced radiosensitivity of CRPC cells both in vitro and in vivo, through EMT reversal [258][42].
2.4. The Role of Tumor Microenviroment (TME)
In addition to the PCa cell itself, several signals arising from the microenvironment can play a key role in governing EMT and highly influence cancer progression or clinical outcomes in PCa patients. A great variety of regulators and inducers, through a complex network of intermingled pathways, facilitate the transition to a less differentiated, aggressive phenotype [72][43]. The key players in the surrounding stroma include vascular and neural networks, fibroblasts (particularly cancer-associated fibroblasts (CAFs)), tumor-associated endothelial cells (TECs), innate and adaptive immune cells, and the altered extracellular matrix [259][44]. EMT changes are not only guided by the tumor cells; they may be induced by stromal cells, which play a leading role not only in tumorigenesis but also in cancer progression and metastasis. Stromal-induced downregulation of miR-1247 by cancer-associated fibroblasts was shown to promote EMT and increased cell invasion, as well as stemness traits in PCa cells [260][45]. In a similar manner, adipose stromal cells (ASC) induce epithelial–mesenchymal transition (EMT) in PCa cells, providing a link between obesity-associated EMT and cancer progression. Interestingly, using a hunter-killer peptide D-CAN, previously developed for targeted ASC, a combination therapy with cisplatin was more effective in suppressing growth of mouse PCa allografts and xenografts, even in nonobese mice [261][46]. Further studies in a genetic model of PCa identified adipose stromal cell-secreted chemocine CXCL12 signaling in prostate epithelium as the main EMT driver and link between obesity and cancer progression [262][47].
Another paradigm of TME-mediated EMT is the Akt/mTOR pathway. In an EMT mouse model, AP1 was found to mediate EMT and the appearance of disseminated tumor cells via the Akt/mTOR pathway [263][48]. Although the exact role of the mTOR pathway in PCa remains unclear, previous studies have underlined p-mTOR expression as a factor influencing lymphangiogenesis and lymph node metastasis in PCa patients [264][49]. A possible link between EMT and the mTOR pathway has been proposed through downregulation of RhoA and Rac1 signaling pathways [265][50]. Interestingly, the use of novel inhibitors, such as the novel dual mTORC1/C2 inhibitor AZD2014, is able to inhibit migration, invasion, and, more importantly, EMT progression in castration-resistant PCa cell lines, suggesting multiple therapeutic benefits from future mTOR inhibitors [266][51]. However, the extremely complex and still undeciphered crosstalk of PCa cells with the ever-adapting microenvironment still poses a challenge in understanding and overcoming TME-induced therapy resistance [259,267][44][52].
(References would be added automatically after the entry is online)
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Torre, L.A.; Siegel, R.L.; Ward, E.M.; Jemal, A. Global Cancer Incidence and Mortality Rates and Trends—An Update. Cancer Epidemiol. Biomark. Prev. 2016, 25, 16–27.
- Sartor, O. Why is prostate cancer incidence rising in young men? Cancer 2020, 126, 17–18.
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386.
- Matuszak, E.A.; Kyprianou, N. Androgen regulation of epithelial–mesenchymal transition in prostate tumorigenesis. Expert Rev. Endocrinol. Metab. 2011, 6, 469–482.
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196.
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428.
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.-J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890.
- Thiery, J.P. Epithelial–mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454.
- Loh, C.-Y.; Chai, J.; Tang, T.; Wong, W.; Sethi, G.; Shanmugam, M.; Chong, P.; Looi, C. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118.
- Pu, H.; Begemann, D.E.; Kyprianou, N. Aberrant TGF-β Signaling Drives Castration-Resistant Prostate Cancer in a Male Mouse Model of Prostate Tumorigenesis. Endocrinology 2017, 158, 1612–1622.
- Røsland, G.V.; Dyrstad, S.E.; Tusubira, D.; Helwa, R.; Tan, T.Z.; Lotsberg, M.L.; Pettersen, I.K.N.; Berg, A.; Kindt, C.; Hoel, F.; et al. Epithelial to mesenchymal transition (EMT) is associated with attenuation of succinate dehydrogenase (SDH) in breast cancer through reduced expression of SDHC. Cancer Metab. 2019, 7, 6.
- Jiborn, T.; Bjartell, A.; Abrahamsson, P.-A. Neuroendocrine Differentiation in Prostatic Carcinoma During Hormonal Treatment. Urology 1998, 51, 585–589.
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to Mesenchymal Transition Contributes to Drug Resistance in Pancreatic Cancer. Cancer Res. 2009, 69, 5820–5828.
- Smith, B.N.; Bhowmick, N.A. Role of EMT in Metastasis and Therapy Resistance. J. Clin. Med. 2016, 5, 17.
- Culig, Z. Epithelial mesenchymal transition and resistance in endocrine-related cancers. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1368–1375.
- Shibue, T.; Weinberg, T.S.R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629.
- Song, B.; Park, S.-H.; Zhao, J.C.; Fong, K.-W.; Li, S.; Lee, Y.; Yang, Y.A.; Sridhar, S.; Lu, X.; Abdulkadir, S.A.; et al. Targeting FOXA1-mediated repression of TGF-β signaling suppresses castration-resistant prostate cancer progression. J. Clin. Investig. 2019, 129, 569–582.
- Shiota, M.; Itsumi, M.; Takeuchi, A.; Imada, K.; Yokomizo, A.; Kuruma, H.; Inokuchi, J.; Tatsugami, K.; Uchiumi, T.; Oda, Y.; et al. Crosstalk between epithelial-mesenchymal transition and castration resistance mediated by Twist1/AR signaling in prostate cancer. Endocr. Relat. Cancer 2015, 22, 889–900.
- Paller, C.; Pu, H.; Begemann, D.E.; Wade, C.A.; Hensley, P.; Kyprianou, N. TGF-β receptor I inhibitor enhances response to enzalutamide in a pre-clinical model of advanced prostate cancer. Prostate 2019, 79, 31–43.
- Shao, C.; Yu, B.; Liu, Y. Androgen receptor splicing variant 7: Beyond being a constitutively active variant. Life Sci. 2019, 234, 116768.
- Ramamurthy, V.P.; Ramalingam, S.; Gediya, L.K.; Njar, V.C.O. The retinamide VNLG -152 inhibits f-AR/AR -V7 and MNK—eIF 4E signaling pathways to suppress EMT and castration-resistant prostate cancer xenograft growth. FEBS J. 2018, 285, 1051–1063.
- Borgmann, H.; Lallous, N.; Ozistanbullu, D.; Beraldi, E.; Paul, N.; Dalal, K.; Fazli, L.; Haferkamp, A.; Lejeune, P.; Cherkasov, A.; et al. Moving Towards Precision Urologic Oncology: Targeting Enzalutamide-resistant Prostate Cancer and Mutated Forms of the Androgen Receptor Using the Novel Inhibitor Darolutamide (ODM-201). Eur. Urol. 2018, 73, 4–8.
- Nam, R.K.; Benatar, T.; Amemiya, Y.; Wallis, C.J.; Romero, J.M.; Tsagaris, M.; Sherman, C.; Sugar, L.; Seth, A. MicroRNA-652 induces NED in LNCaP and EMT in PC3 prostate cancer cells. Oncotarget 2018, 9, 19159–19176.
- Doldi, V.; Pennati, M.; Forte, B.; Gandellini, P.; Zaffaroni, N. Dissecting the role of microRNAs in prostate cancer metastasis: Implications for the design of novel therapeutic approaches. Cell. Mol. Life Sci. 2016, 73, 2531–2542.
- Yeh, Y.; Guo, Q.; Connelly, Z.; Cheng, S.; Yang, S.; Prieto-Domínguez, N.; Yu, X. Wnt/Beta-Catenin Signaling and Prostate Cancer Therapy Resistance. Adv. Exp. Med. Biol. 2019, 1210, 351–378.
- Kolijn, K.; Verhoef, E.I.; Smid, M.; Böttcher, R.; Jenster, G.W.; Debets, R.; van Leenders, G.J. Epithelial–Mesenchymal Transition in Human Prostate Cancer Demonstrates Enhanced Immune Evasion Marked by IDO1 Expression. Cancer Res. 2018, 78, 4671–4679.
- Tsai, Y.-S.; Jou, Y.-C.; Tsai, H.-T.; Cheong, I.-S.; Tzai, T.-S. Indoleamine-2,3-dioxygenase-1 expression predicts poorer survival and up-regulates ZEB2 expression in human early stage bladder cancer. Urol. Oncol. Semin. Orig. Investig. 2019, 37, 810.e17–810.e27.
- Li, F.; Mahato, R.I. MicroRNAs and Drug Resistance in Prostate Cancers. Mol. Pharm. 2014, 11, 2539–2552.
- Puhr, M.; Hoefer, J.; Schäfer, G.; Erb, H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-Mesenchymal Transition Leads to Docetaxel Resistance in Prostate Cancer and Is Mediated by Reduced Expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201.
- Wang, P.; Henning, S.M.; Heber, D.; Vadgama, J.V. Sensitization to docetaxel in prostate cancer cells by green tea and quercetin. J. Nutr. Biochem. 2015, 26, 408–415.
- Lu, X.; Yang, F.; Chen, D.; Zhao, Q.; Chen, D.; Ping, H.; Xing, N. Quercetin reverses docetaxel resistance in prostate cancer via androgen receptor and PI3K/Akt signaling pathways. Int. J. Biol. Sci. 2020, 16, 1121–1134.
- Lu, X.; Chen, D.; Yang, F.; Xing, N. Quercetin Inhibits Epithelial-to-Mesenchymal Transition (EMT) Process and Promotes Apoptosis in Prostate Cancer via Downregulating lncRNA MALAT1. Cancer Manag. Res. 2020, 12, 1741–1750.
- Xue, Y.; Ms, B.Y.; Liu, Y.; Guo, R.; Li, J.; Ms, L.Z.; Su, J.; Sun, L.; Li, Y. Zinc promotes prostate cancer cell chemosensitivity to paclitaxel by inhibiting epithelial-mesenchymal transition and inducing apoptosis. Prostate 2019, 79, 647–656.
- Murata, K.; Saga, R.; Monzen, S.; Tsuruga, E.; Hasegawa, K.; Hosokawa, Y. Understanding the mechanism underlying the acquisition of radioresistance in human prostate cancer cells. Oncol. Lett. 2019, 17, 5830–5838.
- Tsao, T.; Beretov, J.; Ni, J.; Bai, X.; Bucci, J.; Graham, P.; Li, Y. Cancer stem cells in prostate cancer radioresistance. Cancer Lett. 2019, 465, 94–104.
- Deng, X.; Elzey, B.D.; Poulson, J.M.; Morrison, W.B.; Ko, S.-C.; Hahn, N.M.; Ratliff, T.L.; Hu, C.-D. Ionizing radiation induces neuroendocrine differentiation of prostate cancer cells in vitro, in vivo and in prostate cancer patients. Am. J. Cancer Res. 2011, 1, 834–844.
- Chaiswing, L.; Weiss, H.L.; Jayswal, R.D.; Clair, D.K.S.; Kyprianou, N. Profiles of Radioresistance Mechanisms in Prostate Cancer. Crit. Rev. Oncog. 2018, 23, 39–67.
- Wu, X.; Scott, H.; Carlsson, S.V.; Sjoberg, D.D.; Cerundolo, L.; Lilja, H.; Prevo, R.; Rieunier, G.; Macaulay, V.; Higgins, G.S.; et al. Increased EZH2 expression in prostate cancer is associated with metastatic recurrence following external beam radiotherapy. Prostate 2019, 79, 1079–1089.
- Yadav, S.; Kowolik, C.M.; Lin, M.; Zuro, D.; Hui, S.K.; Riggs, A.; Horne, D.A. SMC1A is associated with radioresistance in prostate cancer and acts by regulating epithelial-mesenchymal transition and cancer stem-like properties. Mol. Carcinog. 2019, 58, 113–125.
- Xie, P.; Yu, H.; Wang, F.; Yan, F.; He, X. Inhibition of LOXL2 Enhances the Radiosensitivity of Castration-Resistant Prostate Cancer Cells Associated with the Reversal of the EMT Process. BioMed Res. Int. 2019, 2019, 1–10.
- Montanari, M.; Rossetti, S.; Cavaliere, C.; D’Aniello, C.; Malzone, M.G.; Vanacore, D.; di Franco, R.; La Mantia, E.; Iovane, G.; Piscitelli, R.; et al. Epithelial-mesenchymal transition in prostate cancer: An overview. Oncotarget 2017, 8, 35376–35389.
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68.
- Taddei, M.L.; Cavallini, L.; Ramazzotti, M.; Comito, G.; Pietrovito, L.; Morandi, A.; Giannoni, E.; Raugei, G.; Chiarugi, P. Stromal-induced downregulation of miR-1247 promotes prostate cancer malignancy. J. Cell. Physiol. 2019, 234, 8274–8285.
- Su, F.; Ahn, S.; Saha, A.; DiGiovanni, J.; Kolonin, M.G. Adipose stromal cell targeting suppresses prostate cancer epithelial-mesenchymal transition and chemoresistance. Oncogene 2019, 38, 1979–1988.
- Su, F.; Daquinag, A.C.; Ahn, S.; Saha, A.; Dai, Y.; Zhao, Z.; DiGiovanni, J.; Kolonin, M.G. Progression of prostate carcinoma is promoted by adipose stromal cell-secreted CXCL12 signaling in prostate epithelium. NPJ Precis. Oncol. 2021, 5, 1–10.
- Shi, J.; Wang, L.; Zou, C.; Xia, Y.; Qin, S.; Keller, E.; Mizokami, A.; Zhang, J.; Lu, Y. Tumor microenvironment promotes prostate cancer cell dissemination via the Akt/mTOR pathway. Oncotarget 2018, 9, 9206–9218.
- Lilis, I.; Giopanou, I.; Papadaki, H.; Gyftopoulos, K. The expression of p-mTOR and COUP-TFII correlates with increased lymphangiogenesis and lymph node metastasis in prostate adenocarcinoma. Urol. Oncol. Semin. Orig. Investig. 2018, 36, 311.e27–311.e35.
- Chen, X.; Cheng, H.; Pan, T.; Liu, Y.; Su, Y.; Ren, C.; Huang, D.; Zha, X.; Liang, C. mTOR regulate EMT through RhoA and Rac1 pathway in prostate cancer. Mol. Carcinog. 2015, 54, 1086–1095.
- Li, S.; Sheng, J.; Liu, Z.; Fan, Y.; Zhang, C.; Lv, T.; Hu, S.; Jin, J.; Yu, W.; Song, Y. Potent antitumour of the mTORC1/2 dual inhibitor AZD2014 in docetaxel-sensitive and docetaxel-resistant castration-resistant prostate cancer cells. J. Cell. Mol. Med. 2021, 25, 2436–2449.
- Desbats, M.A.; Giacomini, I.; Prayer-Galetti, T.; Montopoli, M. Metabolic Plasticity in Chemotherapy Resistance. Front. Oncol. 2020, 10, 281.
More