Peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1 α (PGC-1α) is a master transcriptional cofactor for mitochondrial biogenesis. PGC-1α was discovered as a PPARγ-interacting protein that is expressed preferentially in brown adipose tissue (BAT) . PGC-1α binds to transcription factors, such as nuclear respiratory factor (NRF)-1, NRF-2, and estrogen-related receptor α (ERRα), thereby coactivating downstream genes .
- PGC-1α
- obesity
- caloric restriction
- white adipose tissue
1. Introduction
coactivator-1 α (PGC-1α) is a master transcriptional cofactor for mitochondrial biogenesis. PGC-1α binds to transcription factors, such as nuclear respiratory factor (NRF)-1, NRF-2, and estrogen-related receptor α (ERRα), thereby coactivating downstream genes [3,4,5,6][1][2][3][4]. transcriptionally regulate various mitochondrial genes involved in the respiratory chain, and replication and transcription of mitochondrial DNA (mtDNA), which encodes part of proteins comprising respiratory chain complexes [5][3]. Among the downstream mediators of PGC-1α, transcription factor A mitochondria (TFAM) is a major factor responsible for mitochondrial biogenesis [9][5].
Obesity, which is characterized by excess body weight or fat mass due to over-nutrition, causes disturbances in the metabolic, endocrine, and immune systems in the whole body. These obesity-induced abnormalities pose serious health problems, including type 2 diabetes mellitus (T2DM), non-alcoholic fatty liver disease, and cardio- and cerebrovascular diseases Obesity induces many cellular stresses and inflammatory signaling pathways by excess or ectopic accumulation of fat in various tissues, resulting in insulin resistance and hepatic steatosis [16,17][6][7]. Moreover, emerging evidence has indicated a relationship of mitochondrial dysfunction with oxidative stress and systemic inflammation in the obese condition [18][8].
A recent report showed that CR exerted beneficial effects on non-human primates, which suggested the effectiveness of CR in humans [21][9]. Previous studies have described that beneficial effects of CR including various physiological and molecular mechanisms [23][10]. These mechanisms include enhancement of mitochondrial biogenesis, suppression of inflammation, mitigation of oxidative stress, suppression of growth hormone/insulin-like growth factor (GH/IGF-1) signaling, mechanistic target of rapamycin complex 1 activity, and activation of sirtuin. In fact, we have shown that mitochondrial regulation in white adipose tissue (WAT) contributes to the beneficial effects of CR [24,25][11][12].
WAT largely comprises adipocytes, but also comprises adipose-derived stem cells (ADSC), fibroblasts, macrophages, and other immune cells [26][13]. Adipocytes are endocrine cells that secrete adipokines, such as adiponectin, leptin, and pro-inflammatory cytokines [26,27][13][14]. The secretory profile and characteristics of WAT vary depending on the size of adipocytes. In contrast, small adipocytes with a modest amount of TG, which are frequently observed in CR models, secrete more adiponectin and less monocyte chemoattractant protein-1 and TNFα, leading to improved insulin sensitivity in the whole body [36][15].
As mentioned above, mitochondrial regulation is closely implicated in obesity-related pathology and the effects of CR, supporting the relationship between PGC-1α and metabolic states in WAT. Despite many reviews of PGC-1α, few papers currently focus on and comprehensively highlight its link with metabolic states. Therefore, to provide novel insights into the physiological significance of PGC-1α, this review outlines the functions of mitochondria and the involvement of PGC-1α with obesity- and CR-related physiological changes in WAT, which is a tissue involved in whole-body metabolism.
2. Mitochondria and PGC-1α in WAT during Obesity or CR
Adipocytes in WAT contain small and elongated mitochondria in the narrow cytoplasmic space, resulting from a large, unilocular lipid droplet formed by TG [37,38][16][17]. Some previous reports and reviews have described the relationship between mitochondria and white adipocyte-specific functions, such as adipocyte differentiation (or adipogenesis), lipid homeostasis, and insulin sensitivity [39,40][18][19]. Wilson-Fritch and colleagues showed that in 3T3-L1 cells (mouse fibroblasts with the ability to differentiate into adipocytes under specific conditions), mitochondrial capacity is activated depending on differentiation of adipocytes [41][20]. During differentiation of adipocytes, transcriptional factors, including CCAAT/enhancer binding proteins, ERRα, and PPARγ, are sequentially induced, which in turn promote maturation of adipocytes [42,43,44][21][22][23].
Furthermore, mitochondria reportedly provide the key factors for de novo synthesis of both FA and TG, including citrate, glycerol 3-phosphate (G3P), and nicotinamide adenine dinucleotide phosphate (NADPH) [47,48][24][25]. In nutrient-rich conditions, citrate, which is an intermediate of the TCA cycle in mitochondria, is shuttled into the cytosol through the citrate carrier [49][26]. In fact, PGC-1α reportedly induces lipogenesis by the production of citrate in tumors, thereby promoting tumor growth [51][27]. These findings underscore that mitochondria regulate both lipid metabolism and lipogenesis.
Insulin sensitivity is closely involved in lipogenesis via transcriptional regulation of FA synthesis-related factors in white adipocytes [56,57][28][29]. The following studies are examples indicating the association between mitochondria and insulin sensitivity. However, primary adipocytes established from subcutaneous WAT of obese patients have been shown to exhibit increased mitochondrial respiration despite their insulin resistance [59][30]. Therefore, although it remains to be determined whether mitochondria are directly or indirectly associated with insulin sensitivity, mitochondria may participate in lipogenesis via the regulation of insulin signaling in addition to the supply of metabolic intermediates.
Obesity impairs mitochondrial biogenesis and oxidative metabolism in WAT [41][20]. White adipocytes isolated from ob/ob mice or db/db mice (genetic obesity models) show a comprehensive decrease in the expression of genes encoding mitochondrial proteins and a decline in oxygen consumption and citrate synthase activity [42,60,61][21][31][32]. In agreement with the results of experimental animal models, several studies have provided evidence that WAT in humans with obesity shows low levels of mtDNA and proteins comprising mitochondrial respiratory complexes [41,62,63,64][20][33][34][35]. Similarly, human studies have shown that obesity attenuates PGC-1α expression in WAT [63,64,66,67][34][35][36][37].
Adipocytes observed in obese WAT generally show an increased cell size (hypertrophy), rather than cell number (hyperplasia) [41][20]. As described above, hypertrophic adipocytes trigger insulin resistance. Because of the relationship between adipogenesis and mitochondrial biogenesis, mitochondrial regulation by PGC-1α is predicted to be involved in the pathology of obesity via morphological changes in white adipocytes. Therefore, mitochondrial biogenesis regulated by PGC-1α may play an important, but not necessary, role in the differentiation of white adipocytes.
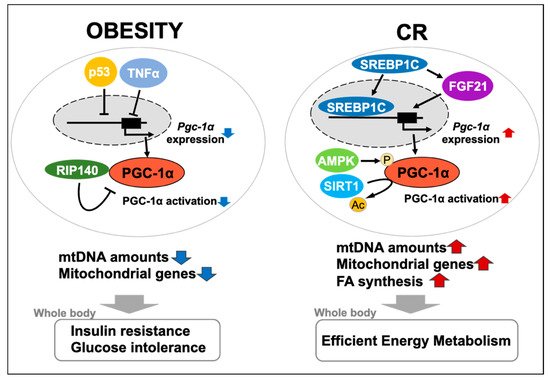
Negative regulators of PGC-1α, including receptor-interacting protein 140 (RIP140), p53, DNA methyltransferase 3 (DNMT3), and MYB binding protein (p160) 1a (MYBBP1a) and Parkin interacting substrate (PARIS), are considered to be responsible for obesity-induced downregulation of PGC-1α [72][38]. Hence, we explain the relationship between each negative regulator and PGC-1α in the following paragraphs.
RIP140 is a coregulator of a number of nuclear receptors and several other transcription factors in various tissues and organs [73,74][39][40]. Subsequently, Powelka and colleagues reported that RIP140 suppressed oxidative metabolism and mitochondrial biogenesis in adipocytes [76][41]. Moreover, RIP140 depletion has been shown to prevent obesity-induced glucose intolerance and insulin resistance in mice [75,76][42][41]. These findings indicate the substantial contribution of RIP140 to the pathology of obesity via downregulated PGC-1α in WAT.
The tumor suppressor p53 is responsive to various stresses, and accumulated evidence has also strongly suggested that p53 greatly contributes to mitochondrial regulation [77,78][43][44]. Our laboratory has identified a suppressive effect of p53 on PGC-1α expression levels and mtDNA content in 3T3-L1 adipocytes, but not in C2C12 myocytes [79][45]. Additionally, Maser and colleagues found that telomere dysfunction-induced p53 repressed PGC-1α expression, which led to mitochondrial dysfunction [80][46]. Similarly, in vivo transient repression of p53 by the CreERT2/loxP system or administration of pifithrin-α (inhibitor of p53 transcriptional activity) induce PGC-1α expression in WAT, and reduce the respiratory exchange ratio [83][47].
DNMT3 plays a central role in epigenetic modifications of the genome via DNA methylation [84][48]. Furthermore, DNMTs are increased in WAT in patients with obesity, thereby inducing the methylation rate of the promoter region of the Krüppel-like factor 4 (KLF4) gene (an anti-inflammatory factor) and suppressing this expression [86][49]. These studies suggest that DNMT3 is associated with obesity-related downregulation of PGC-1α expression and mitochondrial function in adipocytes or WAT, despite no direct evidence. MYBBP1a represses transcriptional activity of PGC-1α by direct interaction in skeletal muscle [87][50].
In fact, many studies have addressed the effects of CR on mitochondrial biogenesis and efficiency in skeletal muscle, brain, or BAT in rodent models [89][51]. In agreement with our result, a microarray study performed by Linford and colleagues showed CR-induced upregulation of genes involved in mitochondrial oxidative phosphorylation in WAT [91][52]. Similarly, Pardo and colleagues showed that CR-induced upregulation of mitochondrial genes in WAT depended on PGC-1α and PGC-1β using double-KO mice [93][53]. Another study also showed that CR was more likely to upregulate mRNA levels of thermogenic genes and Pgc-1α in WAT than in BAT, which represented induction of beiging [94][54].
The suppression of negative regulators of PGC-1α is likely to be a mechanism involved in CR-upregulated PGC-1α expression. Likewise, transcriptome analysis of WAT from CR mice showed p53 gene expression was suppressed by CR [96][55]. Additionally, Wang and colleagues demonstrated that aging induced RIP140 expression, while CR prevented this induction in WAT [97][56]. These studies support the proposal that the suppression of negative regulators can contribute to CR-upregulated PGC-1α expression and mitochondrial biogenesis in WAT.
AMPK enhances the transcriptional activity of PGC-1α by promoting its phosphorylation, and also upregulates Pgc-1α, resulting in activation of mitochondrial metabolism and biogenesis [101,102][57][58]. SIRT1 is involved in mitochondrial biogenesis by stimulating activity of PGC-1α via its deacetylation, despite the controversy over whether this involvement is critical for regulation of PGC-1α [105,106,107,108][59][60][61][62]. SIRT1/PGC-1α axis plays a central role in CR-related regulation of mitochondrial biogenesis in various organs and tissues, and is accepted as a main target of CR mimetics [22][63]. Notably, resveratrol fails to increase mitochondrial gene levels in skeletal muscle of muscle-specific Pgc-1α KO mice, but there are systemic effects of this compound on energy expenditure [111][64].
In addition to the AMPK/SIRT1/PGC-1α axis, we have recently identified sterol regulatory element-binding protein 1c (SREBP1c) and fibroblast growth factor 21 (FGF21) as WAT-specific mediators of CR-induced mitochondrial biogenesis [25,115][12][65]. Our microarray analysis of WAT in ad libitum-fed rats, CR rats, and dwarf rats with suppressed GH/IGF-1 signaling showed that CR induced expression of genes involved in FA synthesis and In CR conditions, Srebp-1c KO mice showed loss of upregulation of genes involved in FA synthesis in WAT and suppression of extension of the lifespan [24][11]. These findings suggest that SREBP-1c is a direct inducer of PGC-1α expression and mitochondrial biogenesis.
However, Fgf21 is expressed in other tissues or organs, such as WAT, BAT, and skeletal muscle [122][66]. Circulating FGF21, which is mostly secreted from the liver, binds to FGF receptor (FGFR) and the beta-klotho (KLB) receptor complex in target tissues, which in turn regulates glucose and lipid metabolism [123][67]. Notably, FGF21 signaling induces Pgc-1α expression in adipocytes [124][68]. We recently reported that CR upregulated Fgf21 and Klb expression, as well as glucose transporter 1 and Pgc-1α expression, which are downstream genes of FGF21 signaling, in rat WATs [115][65].
3. Discussion
Accumulated evidence has shown functional changes of mitochondria in WAT in relation to the systemic metabolic state, including obesity and CR. The cause-and-effect relationship between PGC-1α and the influence of obesity or CR on WAT is complicated. We aimed to provide insight into the physiological significance of alterations in PGC-1α in WAT as follows.
In the early phase of obesity, adipogenesis and lipid anabolism need to be extremely induced to metabolize excess nutrition, probably resulting in over-activation of mitochondria in WAT. The persistence of such a condition in mitochondria is generally considered to trigger production of more reactive oxygen species (ROS), which are byproducts of oxidative phosphorylation. At low levels, ROS play an important role in insulin signal transduction and differentiation of adipocytes [128,129][69][70]. Although PGC-1α is involved in not only mitochondrial biogenesis, but improvement of oxidative stress, obesity-induced downregulation of PGC-1α may represent a defensive reaction to accumulation of abnormal mtDNA
CR is regarded as a chronic and mild energy shortage condition. CR significantly enhances lipid catabolism in the whole body, which allows more efficient systemic metabolism to compensate for an energy shortage [132][71]. A previous study and our study suggest that WAT contributes to this metabolic shift by promoting de novo synthesis of lipids, which are a more efficient source of energy than carbohydrates [25,132][12][71]. Considering that mitochondria play an important role in lipogenesis as mentioned above, CR-induced PGC-1α in WAT may represent enhanced de novo lipogenesis, rather than lipid catabolism by β-oxidation.
In this manuscript, we review findings mainly based on studies that focus on the physiological changes from normal conditions to obese or CR conditions. However, there are many studies that demonstrate CR-related changes in obese conditions [133,134,135,136,137][72][73][74][75][76]. Investigation of the effects of CR on obesity physiology is important for evaluating the clinical significance of CR. Thus, further accumulation of relevant findings will aid in addressing the cause-and-effect relationship between PGC-1α and obesity or CR.
Methodological limitations of the current research mentioned in this review include phenotypic differences between systemic and conditional KO mice. It is conceivable that metabolic alterations are especially susceptible to these differences. For instance, fat-specific conditional KO mice may provide experimental models suitable for more accurate analysis of targeted genes. Hence, the development of a technical method that allows conditional gene deletion to specifically target WAT is desired.
Regulation of mitochondrial dynamics is attributed to not only mitochondrial biogenesis, which is enhanced by PGC-1α, but also mitochondrial degradation by autophagic clearance, namely “mitophagy” [139][77]. Mitophagy is implicated in alterations in characteristics of adipocytes, especially beiging [139][77]. Therefore, a balance of mitochondrial biogenesis and mitophagy is probably important for maintenance of mitochondrial function in WAT, and regulation of PGC-1α may be involved in this balance.
In conclusion, much of the evidence introduced in this review strongly indicates that PGC-1α is a major player in regulating mitochondrial biogenesis or function in WAT in response to a systemic metabolic state (Figure 1). Further investigation of WAT- or white adipocyte-specific function of PGC-1α will lead to identification of the novel mechanisms underlying the relationship between mitochondria and obesity- or CR-related physiological changes.
References
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124.
- Evans, M.J.; Scarpulla, R.C. NRF-1: A trans-activator of nuclearencoded respiratory genes in animal cells. Genes Dev. 1990, 4, 1023–1034.
- Scarpulla, R.C. Nuclear control of respiratory gene expression in mammalian cells. J. Cell Biochem. 2006, 97, 673–683.
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466.
- Virbasius, J.V.; Virbasius, C.A.; Scarpulla, R.C. Identity of GABP with NRF-2, a multisubunit activator of cytochrome oxidase expression, reveals a cellular role for an ETS domain activator of viral promoters. Genes Dev. 1993, 7, 380–392.
- Rutkowski, J.M.; Stern, J.H.; Scherer, P.E. The cell biology of fat expansion. J. Cell. Biol. 2015, 208, 501–512.
- Heymsfield, S.B.; Wadden, T.A. Mechanisms, Pathophysiology, and Management of Obesity. N. Engl. J. Med. 2017, 376, 254–266.
- Prasun, P. Mitochondrial dysfunction in metabolic syndrome. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165838.
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 2017, 8, 14063.
- Hoshino, S.; Kobayashi, M.; Higami, Y. Mechanisms of the anti-aging and prolongevity effects of caloric restriction: Evidence from studies of genetically modified animals. Aging 2018, 10, 2243–2251.
- Fujii, N.; Narita, T.; Okita, N.; Kobayashi, M.; Furuta, Y.; Chujo, Y.; Sakai, M.; Yamada, A.; Takeda, K.; Konishi, T.; et al. Sterol regulatory element-binding protein-1c orchestrates metabolic remodeling of white adipose tissue by caloric restriction. Aging Cell 2017, 16, 508–517.
- Kobayashi, M.; Fujii, N.; Narita, T.; Higami, Y. SREBP-1c-Dependent Metabolic Remodeling of White Adipose Tissue by Caloric Restriction. Int. J. Mol. Sci. 2018, 19, 3335.
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat. Rev. Immunol. 2011, 11, 85–97.
- Cinti, S. The adipose organ. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 9–15.
- Van Baak, M.A.; Mariman, E.C.M. Mechanisms of weight regain after weight loss—The role of adipose tissue. Nat. Rev. Endocrinol. 2019, 15, 274–287.
- Kopecký, J.; Rossmeisl, M.; Flachs, P.; Bardová, K.; Brauner, P. Mitochondrial uncoupling and lipid metabolism in adipocytes. Biochem. Soc. Trans. 2001, 29, 791–797.
- Villarroya, J.; Giralt, M.; Villarroya, F. Mitochondrial DNA: An up-and-coming actor in white adipose tissue pathophysiology. Obesity 2009, 17, 1814–1820.
- De Pauw, A.; Tejerina, S.; Raes, M.; Keijer, J.; Arnould, T. Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am. J. Pathol. 2009, 175, 927–939.
- Heinonen, S.; Jokinen, R.; Rissanen, A.; Pietiläinen, K.H. White adipose tissue mitochondrial metabolism in health and in obesity. Obes. Rev. 2020, 21, e12958.
- Wilson-Fritch, L.; Burkart, A.; Bell, G.; Mendelson, K.; Leszyk, J.; Nicoloro, S.; Czech, M.; Corvera, S. Mitochondrial biogenesis and remodeling during adipogenesis and in response to the insulin sensitizer rosiglitazone. Mol. Cell. Biol. 2003, 23, 1085–1094.
- Lefterova, M.I.; Lazar, M.A. New developments in adipogenesis. Trends Endocrinol. Metab. 2009, 20, 107–114.
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell. Biol. 2006, 7, 885–896.
- Ijichi, N.; Ikeda, K.; Horie-Inoue, K.; Yagi, K.; Okazaki, Y.; Inoue, S. Estrogen-related receptor alpha modulates the expression of adipogenesis-related genes during adipocyte differentiation. Biochem. Biophys. Res. Commun. 2007, 358, 813–818.
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.
- Jitrapakdee, S.; Vidal-Puig, A.; Wallace, J.C. Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cell. Mol. Life Sci. 2006, 63, 843–854.
- Klingenberg, M. Kinetic study of the tricarboxylate carrier in rat liver mitochondria. Eur. J. Biochem. 1972, 26, 587–594.
- Bhalla, K.; Hwang, B.J.; Dewi, R.E.; Ou, L.; Twaddel, W.; Fang, H.B.; Vafai, S.B.; Vazquez, F.; Puigserver, P.; Boros, L.; et al. PGC1α promotes tumor growth by inducing gene expression programs supporting lipogenesis. Cancer Res. 2011, 71, 6888–6898.
- Wong, R.H.; Sul, H.S. Insulin signaling in fatty acid and fat synthesis: A transcriptional perspective. Curr. Opin. Pharmacol. 2010, 10, 684–691.
- Krycer, J.R.; Quek, L.E.; Francis, D.; Zadoorian, A.; Weiss, F.C.; Cookem, K.C.; Nelson, M.E.; Diaz-Vegas, A.; Humphrey, S.J.; Scalzo, R.; et al. Insulin signaling requires glucose to promote lipid anabolism in adipocytes. J. Biol. Chem. 2020, 295, 13250–13266.
- Böhm, A.; Keuper, M.; Meile, T.; Zdichavsky, M.; Fritsche, A.; Häring, H.U.; de Angelis, M.H.; Staiger, H.; Franko, A. Increased mitochondrial respiration of adipocytes from metabolically unhealthy obese compared to healthy obese individuals. Sci. Rep. 2020, 10, 12407.
- Rong, J.X.; Qiu, Y.; Hansen, M.K.; Zhu, L.; Zhang, V.; Xie, M.; Okamoto, Y.; Mattie, M.D.; Higashiyama, H.; Asano, S.; et al. Adipose mitochondrial biogenesis is suppressed in db/db and high-fat diet-fed mice and improved by rosiglitazone. Diabetes 2007, 56, 1751–1760.
- Choo, H.J.; Kim, J.H.; Kwon, O.B.; Lee, C.S.; Mun, J.Y.; Han, S.S.; Yoon, Y.S.; Yoon, G.; Choi, K.M.; Ko, Y.G. Mitochondria are impaired in the adipocytes of type 2 diabetic mice. Diabetologia 2006, 49, 784–791.
- Pietiläinen, K.H.; Naukkarinen, J.; Rissanen, A.; Saharinen, J.; Ellonen, P.; Keränen, H.; Suomalainen, A.; Götz, A.; Suortti, T.; Yki-Järvinen, H.; et al. Global transcript profiles of fat in monozygotic twins discordant for BMI: Pathways behind acquired obesity. PLoS Med. 2008, 5, e51.
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145.
- Heinonen, S.; Muniandy, M.; Buzkova, J.; Mardinoglu, A.; Rodríguez, A.; Frühbeck, G.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Kaprio, J.; et al. Mitochondria-related transcriptional signature is downregulated in adipocytes in obesity: A study of young healthy MZ twins. Diabetologia 2017, 60, 169–181.
- Semple, R.K.; Crowley, V.C.; Sewter, C.P.; Laudes, M.; Christodoulides, C.; Considine, R.V.; Vidal-Puig, A.; O’Rahilly, S. Expression of the thermogenic nuclear hormone receptor coactivator PGC-1alpha is reduced in the adipose tissue of morbidly obese subjects. Int. J. Obes. Relat. Metab. Disord. 2004, 28, 176–179.
- Hammarstedt, A.; Jansson, P.A.; Wesslau, C.; Yang, X.; Smith, U. Reduced expression of PGC-1 and insulin-signaling molecules in adipose tissue is associated with insulin resistance. Biochem. Biophys. Res. Commun. 2003, 301, 578–582.
- Wenz, T. Regulation of mitochondrial biogenesis and PGC-1α under cellular stress. Mitochondrion 2013, 13, 134–142.
- Nautiyal, J.; Christian, M.; Parker, M.G. Distinct functions for RIP140 in development, inflammation, and metabolism. Trends Endocrinol. Metab. 2013, 24, 451–459.
- Nautiyal, J. Transcriptional coregulator RIP140: An essential regulator of physiology. Mol. Endocrinol. 2017, 58, R147–R158.
- Powelka, A.M.; Seth, A.; Virbasius, J.V.; Kiskinis, E.; Nicoloro, S.M.; Guilherme, A.; Tang, X.; Straubhaar, J.; Cherniack, A.D.; Parker, M.G.; et al. Suppression of oxidative metabolism and mitochondrial biogenesis by the transcriptional corepressor RIP140 in mouse adipocytes. J. Clin. Investig. 2006, 116, 125–136.
- Leonardsson, G.; Steel, J.H.; Christian, M.; Pocock, V.; Milligan, S.; Bell, J.; So, P.W.; Medina-Gomez, G.; Vidal-Puig, A.; White, R.; et al. Nuclear receptor corepressor RIP140 regulates fat accumulation. Proc. Natl. Acad. Sci. USA 2004, 101, 8437–8442.
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622.
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292.
- Okita, N.; Ishikawa, N.; Mizunoe, Y.; Oku, M.; Nagai, W.; Suzuki, Y.; Matsushima, S.; Mikami, K.; Okado, H.; Sasaki, T.; et al. Inhibitory effect of p53 on mitochondrial content and function during adipogenesis. Biochem. Biophys. Res. Commun. 2014, 446, 91–97.
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365.
- Fu, W.; Liu, Y.; Sun, C.; Yin, H. Transient p53 inhibition sensitizes aged white adipose tissue for beige adipocyte recruitment by blocking mitophagy. FASEB J. 2019, 33, 844–856.
- Loaeza-Loaeza, J.; Beltran, A.S.; Hernández-Sotelo, D. DNMTs and Impact of CpG Content, Transcription Factors, Consensus Motifs, lncRNAs, and Histone Marks on DNA Methylation. Genes 2020, 11, 1336.
- Deng, Y.; Qiu, T.; Zhang, M.; Wu, J.; Zhang, X.; Wang, J.; Chen, K.; Feng, J.; Ha, X.; Xie, J.; et al. High Level of Palmitic Acid Induced Over-Expressed Methyltransferase Inhibits Anti-Inflammation Factor KLF4 Expression in Obese Status. Inflammation 2020, 43, 821–832.
- Fan, M.; Rhee, J.; St-Pierre, J.; Handschin, C.; Puigserver, P.; Lin, J.; Jäeger, S.; Erdjument-Bromage, H.; Tempst, P.; Spiegelman, B.M. Suppression of mitochondrial respiration through recruitment of p160 myb binding protein to PGC-1alpha: Modulation by p38 MAPK. Genes Dev. 2004, 18, 278–289.
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532.
- Linford, N.J.; Beyer, R.P.; Gollahon, K.; Krajcik, R.A.; Malloy, V.L.; Demas, V.; Burmer, G.C.; Rabinovitch, P.S. Transcriptional response to aging and caloric restriction in heart and adipose tissue. Aging Cell 2007, 6, 673–688.
- Pardo, R.; Vilà, M.; Cervela, L.; de Marco, M.; Gama-Pérez, P.; González-Franquesa, A.; Statuto, L.; Vilallonga, R.; Simó, R.; Garcia-Roves, P.M.; et al. Calorie restriction prevents diet-induced insulin resistance independently of PGC-1-driven mitochondrial biogenesis in white adipose tissue. FASEB J. 2019, 33, 2343–2358.
- Sheng, Y.; Xia, F.; Chen, L.; Lv, Y.; Lv, S.; Yu, J.; Liu, J.; Ding, G. Differential Responses of White Adipose Tissue and Brown Adipose Tissue to Calorie Restriction During Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 393–399.
- Derous, D.; Mitchell, S.E.; Green, C.L.; Wang, Y.; Han, J.D.J.; Chen, L.; Promislow, D.E.L.; Lusseau, D.; Douglas, A.; Speakman, J.R. The Effects of Graded Levels of Calorie Restriction: X. Transcriptomic Responses of Epididymal Adipose Tissue. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 279–288.
- Wang, J.; Chen, X.; Osland, J.; Gerber, S.J.; Luan, C.; Delfino, K.; Goodwin, L.; Yuan, R. Deletion of Nrip1 Extends Female Mice Longevity, Increases Autophagy, and Delays Cell Senescence. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 882–892.
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022.
- Garcia-Roves, P.M.; Osler, M.E.; Holmström, M.H.; Zierath, J.R. Gain-of-function R225Q mutation in AMP-activated protein kinase gamma3 subunit increases mitochondrial biogenesis in glycolytic skeletal muscle. J. Biol. Chem. 2008, 283, 35724–35734.
- Nemoto, S.; Fergusson, M.M.; Finkel, T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J. Biol. Chem. 2005, 280, 16456–16460.
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118.
- Gerhart-Hines, Z.; Rodgers, J.T.; Bare, O.; Lerin, C.; Kim, S.H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. EMBO J. 2007, 26, 1913–1923.
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95.
- Madeo, F.; Carmona-Gutierrez, D.; Hofer, S.J.; Kroemer, G. Caloric Restriction Mimetics against Age-Associated Disease: Target, Mechanisms, and Therapeutic Potential. Cell Metab. 2019, 29, 592–610.
- Svensson, K.; Schnyder, S.; Albert, V.; Cardel, B.; Quagliata, L.; Terracciano, L.M.; Handschin, C. Resveratrol and SRT1720 Elicit Differential Effects in Metabolic Organs and Modulate Systemic Parameters Independently of Skeletal Muscle Peroxisome Proliferator-activated Receptor γ Co-activator 1α (PGC-1α). J. Biol. Chem. 2015, 290, 16059–16076.
- Fujii, N.; Uta, S.; Kobayashi, M.; Sato, T.; Okita, N.; Higami, Y. Impact of aging and caloric restriction on fibroblast growth factor 21 signaling in rat white adipose tissue. Exp. Gerontol. 2019, 118, 55–64.
- Fon Tacer, K.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 2010, 24, 2050–2064.
- Kilkenny, D.M.; Rocheleau, J.V. The FGF21 Receptor Signaling Complex: Klothoβ, FGFR1c, and Other Regulatory Interactions. Vitam. Horm. 2016, 101, 17–58.
- Chau, M.D.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558.
- Loh, K.; Deng, H.; Fukushima, A.; Cai, X.; Boivin, B.; Galic, S.; Bruce, C.; Shields, B.J.; Skiba, B.; Ooms, L.M.; et al. Reactive oxygen species enhance insulin sensitivity. Cell Metab. 2009, 10, 260–272.
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544.
- Bruss, M.D.; Khambatta, C.F.; Ruby, M.A.; Aggarwal, I.; Hellerstein, M.K. Calorie restriction increases fatty acid synthesis and whole body fat oxidation rates. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E108–E116.
- Aksungar, F.B.; Sarıkaya, M.; Coskun, A.; Serteser, M.; Unsal, I. Comparison of Intermittent Fasting Versus Caloric Restriction in Obese Subjects: A Two Year Follow-Up. J. Nutr. Health Aging 2017, 21, 681–685.
- Tam, C.S.; Covington, J.D.; Ravussin, E.; Redman, L.M.; Pennington CALERIE Team. Little evidence of systemic and adipose tissue inflammation in overweight individuals. Front. Genet. 2012, 3, 58.
- Bussey, C.E.; Withers, S.B.; Aldous, R.G.; Edwards, G.; Heagerty, A.M. Obesity-Related Perivascular Adipose Tissue Damage Is Reversed by Sustained Weight Loss in the Rat. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1377–1385.
- Rodrigues, M.O.M.; Evangelista-Silva, P.H.; Neves, N.N.; Moreno, L.G.; Santos, C.S.; Rocha, K.L.S.; Ottone, V.O.; Batista-da-Silva, B.; Dias-Peixoto, M.F.; Magalhães, F.C.; et al. Caloric restriction-induced weight loss with a high-fat diet does not fully recover visceral adipose tissue inflammation in previously obese C57BL/6 mice. Appl. Physiol. Nutr. Metab. 2020, 45, 1353–1359.
- La Russa, D.; Marrone, A.; Mandalà, M.; Macirella, R.; Pellegrino, D. Antioxidant/Anti-Inflammatory Effects of Caloric Restriction in an Aged and Obese Rat Model: The Role of Adiponectin. Biomedicines 2020, 8, 532.
- Altshuler-Keylinand, S.; Kajimura, S. Mitochondrial homeostasis in adipose tissue remodeling. Sci. Signal. 2017, 10, eaai9248.