Metabolic changes in tumor microenvironment play a critical role in cancer, related to the accumulated alterations in signaling pathways that control cellular metabolism. Cancer metabolic deregulation is related to specific events such as the control of oxidative stress, and in particular the redox imbalance with aberrant oxidant production and/or a deregulation of the efficacy of the antioxidant systems. In cancer cells, different cytokines are involved in the development and/or progression of cancer: among these cytokines, the transforming growth factor β (TGF-β) is central to tumorigenesis and cancer progression. In tumor cells, it has been demonstrated that there is a close correlation between oxidative stress and TGF-β: this crosstalk strongly contributes to tumorigenesis, both in tumor development and in mediating its invasiveness.
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Cancer metabolism involves different changes at cellular level, and altered metabolic pathways have been demonstrated to be involved in tumorigenesis and invasiveness.
Metabolic changes in the tumor microenvironment play a critical role in cancer, especially in the uncontrolled cell proliferation related to accumulated alterations in the signaling pathways that control cellular metabolism, which in turn sustain enhanced cell proliferation. Thus, to achieve and sustain that proliferative capacity, cancer cells must induce or modulate some metabolic pathways
[1].
Cancer metabolic deregulation is related to specific events such as the control of oxidative stress, in particular the redox imbalance with aberrant oxidant production and/or a lower or higher efficacy of antioxidant systems, and the epithelial to mesenchymal transition (EMT)
[2][3][2,3], a physiological event widely involved in tumor development and metastases. Both of these events are differently linked to the cytokine transforming growth factor
β (TGF-
β), whose role in tumorigenesis has been extensively studied
[4], although it has not yet been fully clarified; there is a close correlation between TGF-
β and oxidative stress, a strong crosstalk that can contribute to the development and/or progression of the tumor.
2. Transforming Growth Factor β (TGF-β) in Cancer Metabolism
In cancer cells, different cytokines are involved in the development and/or progression of cancer. Among these cytokines, TGF-
β is central to tumorigenesis and cancer progression, although depending on the cancer stage
[4].
TGF-
β belongs to a large family of more than 40 proteins, which are organized in several subfamilies. Among the different TGF-
β isoforms, at least three genetically distinct ones are particularly expressed, called TGF-
β1, TGF-
β2, and TGF-
β3, with a high homology, but collectively referred to as TGF-
β. TGF-
β binds to its receptor on the plasma membrane and activates downstream signaling by phosphorylation of different mediators, mainly SMAD proteins, which are the main effector molecules in the TGF-
β signaling pathway
[5]. The interaction between SMAD and other downstream proteins in turn controls the expression of specific target genes and the regulation of nuclear or cytoplasmic proteins. An alternative pathway, not mediated by SMAD, is also known
[6], and it has been demonstrated to involve in particular the GTPases Ras and Rho and the mitogen-activated protein kinases (MAPK), all of which have been widely shown to be activated by TGF-
β [7].
TGF-
β has been demonstrated to have a dual role in tumor progression: on the one hand, TGF-
β acts as a tumor suppressor, especially in the early stages of tumor development, where TGF-
β promotes apoptosis and inhibits the proliferation of cancer cells
[4]. On the other hand, in the advanced stage of the tumor, TGF-
β has a pro-tumorigenic effect, promoting genomic mutations and events associated with the malignant progression of cancer, such as EMT (Epithelial Mesenchymal Transition), angiogenesis, inhibition of the immune response, cell motility, and thus the promotion of metastases
[4][8][4,8]. At a DNA level, many genetic and epigenetic alterations have been associated in different types of cancer with variations in the TGF-
β signaling, both in suppression and tumor promotion. At a clinical level, in particular, TGF-
β expression is significantly increased during tumor progression, and this event often is correlated to a poor prognosis
[4].
Furthermore, it seems that during tumor progression TGF-
β acts differently, not only in correlation to the stage in carcinogenesis, but also according to the different reactivity of the tumor cells: this response may be associated with various factors, independently or related to TGF-
β and its receptor expression, the availability of downstream signaling components, evasion of the immune response, stimulation of inflammation, and recruitment of cells that promote tumor growth
[4]. Above all, some studies have shown that TGF-
β expression is often increased in cell lines and tumor tissues compared to normal cells or tissues, while other studies have shown that the growth inhibition induced by TGF-
β in non-transformed cells is often impaired in carcinomas
[9], and in response to TGF-
β, normal cells increase migration and invasiveness, especially via EMT, thus promoting cancer progression
[10].
Therefore, several tumors express high levels of TGF-
β, and this event correlates with a greater aggressiveness of the tumor and consequently a poor clinical prognosis
[9]. Some alterations of key components involved in TGF-
β signaling have been identified in association with oncogenic mutations, among which are those affecting SMAD, particularly the SMAD4 gene
[11], which has been shown to be the most frequent alteration playing a key role in carcinogenesis, as in pancreatic ductal adenocarcinoma and colorectal and gastrointestinal cancers
[12][13][14][15][12,13,14,15]. Finally, TGF-
β acts as a potent immunosuppressive cytokine in cancer cells, enabling these cells to escape the surveillance exerted by the immune system, thereby promoting tumor growth and metastasis
[16].
32. TGF-β and Oxidative Stress Crosstalk in Cancer Cell Metabolism
It has been widely demonstrated that TGF-
β is able to regulate ROS (Reactive Oxygen Species) levels; on the one hand by increasing its production, on the other hand by inhibiting the activity of antioxidant or scavenging systems at a cellular level
[17][18][52,53]. Moreover, the increased level of ROS can in turn induce the expression of TGF-
β and stimulate its release, thus making it active
[17][19][52,54]. At the same time, it is known that ROS are mediators of many effects exerted by TGF-
β, which in turn can modulate ROS levels by increasing their production and reducing the activity of antioxidant systems.
In tumor cells, it has been demonstrated that there is a close correlation between oxidative stress and TGF-
β (
Figure 1). ROS has been shown to mediate many effects of TGF-
β in cancer progression, by deregulating tumor suppressors and, at the same time, by promoting tumorigenesis; one of the mechanisms of crosstalk between ROS and TGF-
β in cancer cell metabolism shows ROS as directly involved in the regulation of the TGF-
β pathway, thus involving factors such as SMAD, MAPK and NF-kB, and in turn promoters of cell proliferation and motility
[20][55], particularly by increasing SMAD and thus making tumor cells resistant to the inhibition of proliferation exerted by TGF-
β in the initial step of tumorigenesis
[21][56]. Furthermore, in cancer cells TGF-
β is involved in many other signaling pathways, such as PI3K/Akt or MAPK, which, in turn, regulate redox-sensitive transcription factors, such as NF-kB or HIF-1
α [22][57].
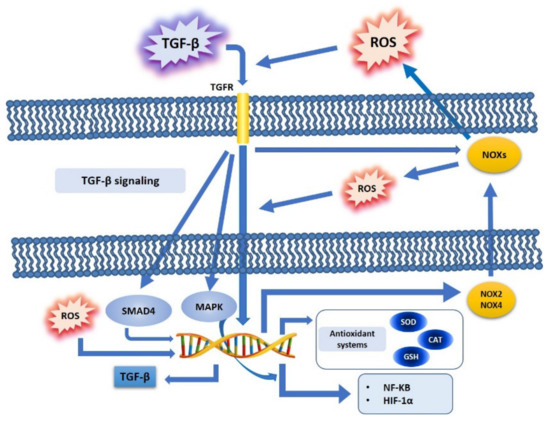
Figure 1. TGF-β and ROS crosstalk. TGF-β binds its cell surface receptors and induces the activation of downstream pathways (SMAD, MAPK), and further regulates ROS production (NOXs activation, NOX 2 and 4), or modulates antioxidant systems (SOD, CAT, GSH) and redox-sensitive transcription factors (NF-kB, HIF-1α). Furthermore, increased ROS production may directly induce TGF-β expression at a nuclear level.
32.1. TGF-β, ROS, and NOX Family
The NOX (NADPH oxidase) family plays an important role in mediating the actions of TGF-
β via the ROS produced, and this effect is found to be deregulated in tumors. In particular, ROS generated by NOX4 mediate apoptosis, which in turn is mediated by the TGF-
β signaling pathway
[23][58]: it has been shown that NOX4 can mediate TGF-
β effects, while NOX-dependent redox signaling can regulate TGF-
β signaling. TGF-
β activates NOXs via Rac1
[24][59]: it has been shown that this factor induces NOX4 in different cell types, both
in vivo and
in vitro [17][52]. However, the induction of NOX4 gene expression by TGF-
β particularly occurs via SMAD3, and this effect has been shown in breast cancer cells
[25][60]. Furthermore, TGF-
β has been demonstrated to induce NOX4 gene expression in crosstalk with an increase in ROS production, while NOX4 is downregulated with reduced ROS synthesis, thus indicating, in pancreatic cancer cells, that NOX4 is the main source of ROS
[26][61]. Finally, it has been shown, in adenocarcinoma Hela cells, that TGF-
β can also induce NOX2 gene expression and its activation
[27][62] (
Figure 1).
TGF-
β induces and activates NOXs with different effects, depending on the member of the NOX family. NOX4 induction by TGF-
β may mediate some of its suppressor effects, such as apoptosis or senescence: it has been shown that TGF-
β induces senescence in hepatocellular carcinoma cells and inhibits tumor growth
[28][63]. This mechanism has been explained as mediated by the liver-specific tumor suppressor STAT5, which in turn controls NOX4 expression and, consequently, drives the proapoptotic proteins PUMA and BIM in mice
[29][64], as NOX4 upregulation via TGF-
β in hepatocytes is required for its pro-apoptotic activity
[30][65]. On the contrary, other members of the family, dependent on Rac1 and activated by TGF-
β, might have opposite effects
[24][59], such as inhibition of the epidermal growth factor (EGF) pathway, which enhances TGF-
β induced apoptosis in rat hepatoma cells, thus inducing oxidative stress via increased ROS production, coincident with a change in the expression pattern of the NOX isoforms
[31][32][66,67].
32.2. TGF-β, ROS, and Antioxidant Systems
TGF-
β is able to increase ROS levels by downregulating the expression of antioxidant systems. TGF-
β regulates the activity of ROS, both by modulating their production and by downregulating the expression of antioxidant enzymes, such as SOD and CAT
[17][33][52,68], and by modulating cellular antioxidant molecules, such as GSH
[17][52].
Concerning SOD and CAT, it has been shown that these antioxidant enzymes are downregulated in many tumors
[34][35][32,36], and some papers have demonstrated that TGF-
β is the mediator in suppressing the expression of Mn-SOD and CAT
[33][68].
It was shown that TGF-
β induced a decrease in the concentration of GSH, inhibiting the expression of the catalytic subunit of the gamma-glutamylcysteine synthetase (GLC) enzyme: TGF-
β, by inhibiting GLC expression, causes a strong reduction of GLC activity, thus also promoting GSH inhibition in the human alveolar epithelial cells of lung adenocarcinoma
[36][69] (
Figure 1).
32.3. TGF-β, ROS, and Redox-Sensitive Factors
TGF-β is linked to NF-kB in a ROS dependent way
[24][59]: TGF-
β activates NF-kB in a ROS dependent manner, and in turn ROS modulate the NF-kB signaling pathway, especially by increasing the IKK/NEMO dimerization, an event mediated by ROS-sensitive IKK phosphatases. In this way, active IKK phosphorylates IKB, thus inducing nuclear translocation and activation of NF-kB
[37][47] (
Figure 1).
Concerning HIF-1
α, it has been shown that this factor induces TGF-
β production, and that, at the same time, hypoxia stabilizes HIF-1
α, mainly by promoting EMT and related cancer metastasis
[38][70]: ROS are not only produced by an aberrant function of mitochondrial complex III during hypoxic stress, but ROS are also stabilized by HIF-1
α, thus interacting with Snail during EMT
[20][55], in order to enhance the invasiveness in cancer cells
[39][71] (
Figure 1).
32.4. TGF-β, ROS, and the Epithelial Mesenchymal Transition (EMT)
The EMT is an event characterized on the one hand by the loss of polarity and cell junctions of the epithelial cells, and, on the other hand, by an increased cellular motility and the acquisition of a mesenchymal phenotype, which gives rise to embryonic development and tissue reconstruction under normal physiological conditions. However, this process can be pathological, as EMT drives fibrosis development and cancer. It is commonly accepted that TGF-
β-induced EMT is a key step in mediating tumor invasiveness and metastasis
[40][72].
TGF-
β induces the EMT of transformed cells, and this event makes the tumor capable of invading surrounding tissues and promoting metastases. In fact, EMT represents one of the key steps that drive the acquisition of a metastatic phenotype in cancer cells, strongly collaborating in the pathogenesis of cancer and the tumor microenvironment
[41][73]. The ability of TGF-
β to induce EMT is a crosstalk with oxidative stress: ROS synergize with TGF-
β both in the initiation and progression of cancer via EMT, not only by increasing the redox imbalance in tumor cells, but also by co-inducing EMT, which is then hyperactivated in carcinoma cells
[42][74].
Therefore, it has been shown that TGF-
β and ROS participate in inducing EMT: several studies have demonstrated that, in tumor cells incubated with antioxidant treatments, the ability of TGF-
β to induce EMT is decreased, suggesting the crucial role exerted by oxidative stress. Furthermore, ROS production, whether TGF-
β-dependent or not, can increase TGF-
β expression, which in turn may contribute to the development of EMT
[43][75].
While the link between carcinogenesis and oxidative stress, through chronic inflammation, has found support in research studies on a wide range of tissue types, the link between ROS and EMT is less well defined. ROS produced at a mitochondrial level, via MMP-3/Rac1b, can induce EMT with loss of E-cadherin and an increase of vimentin and Snail, and with a consequent increase in invasiveness and motility
[44][76]. Cannito et al.
[45][77] demonstrated that ROS are crucial in the early stages of EMT, in the crosstalk with hypoxia, in hepatoblastoma, colon, and pancreas cell lines
[45][77]: it has been shown that the EMT induced by hypoxia is related to an increased intracellular ROS production, which in turn leads to GSK-3
β phosphorylation and inactivation, and in turn is linked to the SMAD-mediated TGF-
β pathway. Recently, it has been demonstrated in human epithelial cells that low doses of H
2O
2 induce EMT by downregulation of E-cadherin and Zonula Occludens 1 expression, with a simultaneous upregulation of α-SMA, and by promoting EMT
[46][78].
The control of cellular redox homeostasis appears to be related to EMT, and oxidative stress is an important factor that increases tumor malignancy
[47][79]. Oxidative stress can in turn modify the cellular response to TGF-
β, in a growing crosstalk that increasingly leads cancer cells to be aggressive and more invasive. This TGF-
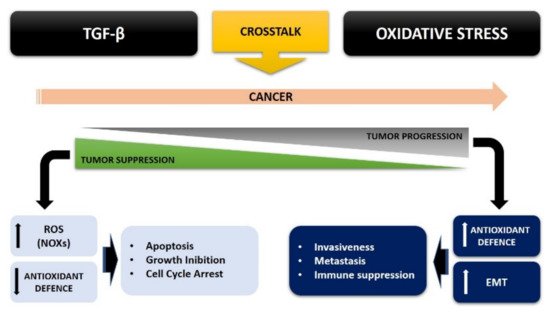
β overexpression may in turn be regulated by ROS production within the cancer, via both a SMAD and non-SMAD signaling pathway, leading to EMT (
Figure 2).
Figure 2. TGF-β and oxidative stress crosstalk in cancer cells.
2.5. TGF-β and ROS Crosstalk in Cancer: A Possible Therapeutic Approach
To date, several studies have been undertaken seeking a possible therapeutic approach to cancer by exploiting the studies done on oxidative stress and the TGF-
β pathway. In recent years, some antioxidants (vitamins C and E) have been proposed as potential drugs able to counteract oxidative stress in cancer development
[48][80]. However, most antioxidants taken orally have limited absorption profile, which leads to low bioavailability and insufficient concentrations at the target site
[48][49][80,81]. Therefore, some studies have performed experiments with specific nanoparticles with intrinsic antioxidant properties and also functionalized for targeted therapy. Finally, the use of antioxidants, such as N-acetylcysteine or NOX inhibitors, in cancer cells has been demonstrated to be efficient in inhibiting cell proliferation, invasion, and metastasis
[48][49][80,81].
Similarly, other studies have identified the TGF-
β pathway as a target for an anti-cancer pharmacological approach, particularly concerning the role of TGF-
β in mediating the immunosuppression of immune cells and promoting angiogenesis and EMT in cancer cells
[4]. It has been showed that a T cell specific blockade of TGF-
β signaling could enhance anti-tumor immunity via a cytotoxic T cell response
[50][82], while recently, some studies have demonstrated that the overexpression of E-cadherin, the main epithelial marker of EMT downregulated in cancer cells, could suppress cellular migration and invasiveness, while the inhibition of N-cadherin, a mesenchymal marker of EMT overexpressed in cancer cells, caused the opposite effects
[51][83].
However, it would be useful to combine these different therapeutic approaches in cancer treatment, in an attempt, given the clear crosstalk between oxidative stress and TGF-
β, to improve cancer treatment.
3. Conclusions
Therefore, the crosstalk between ROS and TGF-β strongly contributes to tumorigenesis, thus bypassing the inhibition of cell proliferation and increasing the malignancy of tumor cells.
So, this ROS/TGF-β crosstalk strongly contributes to tumorigenesis, by avoiding the inhibition of cell proliferation and increasing the malignancy of tumor cells, where oxidative stress can convert the antitumor role of TGF-β in early tumor progression into a pro-tumorigenic role, thus signaling a malignant phenotype of cancer.
Take as a whole, it is crucial to better understand this crosstalk between TGF-
β and oxidative stress in cancer cell metabolism, in an attempt to improve the pharmacological and therapeutic approach against cancer, and also considering the growing interest in clarifying this important topic.