Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Giuseppina Basta and Version 2 by Rita Xu.
The interaction between the membrane spike (S) protein of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and the transmembrane angiotensin-converting enzyme 2 (ACE2) receptor of the human epithelial host cell is the first step of infection, which has a critical role for viral pathogenesis of the current coronavirus disease-2019 (COVID-19) pandemic.
- COVID-19
- SARS-CoV-2
- protease
- ACE2
- repositioning drugs
- co-receptors
1. Introduction
Over the last two decades, there have been three deadly human outbreaks of coronaviruses (CoV), severe acute respiratory syndrome-CoV (SARS-CoV), Middle East Respiratory Syndrome-CoV (MERS-CoV), and SARS-CoV-2. The latter is causing the current pandemic called CoV disease 2019 (COVID-19). They target the human respiratory tract causing severe progressive pneumonia and could spread to other organs, causing damage to the central nervous system in SARS-CoV, severe renal failure in MERS-CoV, and multi-organ failure in SARS-CoV-2 [1].
Despite a high percentage of people with a positive screening test results asymptomatic or paucisymptomatic, COVID-19 can manifest as a respiratory tract infection with a serious spectrum of infection [2]. Severe symptoms, with hypoxia and pneumonia was reported in 15 to 20 percent of infections [3], with a critical associated acute respiratory distress syndrome (ARDS), which can rapidly progress to a multi-organ failure, irreversible and lethal in some cases [4][5][4,5]. Genomic studies confirmed the role of viral spike glycoprotein (S protein) in virulence and pathogenicity for SARS-CoV, MERS-CoV and SARS-CoV-2 [1].
The inflammatory cascade, fibrotic and coagulative events of COVID-19 start from the interaction between the membrane S protein of SARS-CoV-2 and the transmembrane angiotensin-converting enzyme 2 (ACE2) used as site of attachment to the host cell. However, its entry into the host cells is mediated by transmembrane proteases, of which the transmembrane serine protease 2 (TMPRSS2) is the main one. Recent studies have identified several key amino-acidic residues for S-protein interactions with the human ACE2 receptor and the TMPRSS2 membrane protease to initiate infection [6].
Although ACE2 is a target receptor for both SARS-CoV and SARS-CoV-2, the genetic variance observed in the homologous sequence of the gene encoding the S protein allows SARS-CoV-2 to bind efficiently to the receptor with firm attachment, improving virulence compared to SARS-CoV, and then causing very high morbidity and mortality worldwide.
Since ACE2 and TMPRSS2 are co-expressed in a limited number of tissues, the high viral transmissibility and the tissue tropism suggest that SARS-CoV-2 may use other proteases for cellular entry [7]. In fact, several proteases have been found to be involved in the transmission or infection process, including furin (a membrane-bound protease expressed in different tissues, mainly in the lungs [6]), ADAM17 (short for a disintegrin and metalloprotease 17), and cathepsin L.
Several studies have also focused on identifying additional mediators which may increase SARS-CoV-2 infectivity and contribute to the tissue/organ tropism. Some data are emerging for other cell mediators/receptors, including neuropilin-1 (NRP-1), integrins, sialic acids (SA), factor Xa, heparan sulfate (HS), cluster of differentiation 147 (CD147) and glucose-regulated protein 78 (GRP78) [8].
Given the complexity of interactions between viral proteins and host receptors with differing binding specificity and affinity, the differential prognosis for COVID-19 in SARS-CoV-2 positive patients may depend on the presence of single-nucleotide polymorphism in ACE2, serine proteases, mediators or co-receptors, either individually or combined with each other or even in combination with SARS-CoV-2 genetic variants resulting in more or less virulent and lethal strains [9].
To date, effective antivirals for counteract COVID-19 have not been found and many bio-molecular mechanisms of SARS-CoV-2 infection remain elusive.
The identification of key factors, such as receptors and proteases, involved in the dynamic of infection, could provide a way to stop the spread of the virus and suggest single or combined therapeutic treatments to counteract host binding and multi-protease activation.
2. The Binding of SARS-CoV-2 to ACE2 Receptor Is Just the Tip of the Iceberg: The Submerged Front of Multi-Proteases
Both SARS-CoV and SARS-CoV-2 utilize ACE2 as a host-cell entry receptor and proteases as entry activators [10] (Table 1). ACE2 is a negative regulator of the renin–angiotensin system (RAS), which is essential for maintaining blood pressure homeostasis and the balance of salts and fluid. This regulation is critical for the physiopathology of various organs, including lungs, kidneys, and heart. ACE2 also regulates the absorption of amino acids in the gut and kidney, then modulating the expression of transporters for amino acids [10][11][10,11].
Table 1. Receptors, co-receptors and proteases involved in the infection of the three recent coronavirus outbreaks.
| SARS-CoV-2 | SARS-CoV | MERS-CoV | |
|---|---|---|---|
| Receptor | ACE2 [10] | ACE2 [12] | DPP4 (CD26) [10] |
| Priming protease | TMPRSS2 [13] | TMPRSS2 [12] | |
| Furin [14] | Furin [15] | TMPRSS2 [16] | |
| Cathepsin-L [16] | Cathepsin-L [17] | Furin [16] | |
| ADAM17 [17] | ADAM 17 [12] | Cathepsin-L [18] | |
| Factor Xa [19] | Factor Xa [20] | ||
| Co-receptors | Sialic Acids [21] | ||
| NRP-1 [22] | Integrins [23] | Sialic Acids [21] | |
| Integrins [24] | Heparan Sulfate [25] | ||
| Heparan Sulfate [26] | |||
| Other receptors | CD147 [27] | CD147 [17] | GRP78 [28] |
| GRP78 [28] | GRP78 [28] |
ACE2: angiotensin-converting enzyme 2; DPP4; dipeptidyl peptidase-4; TMPRSS2: transmembrane serine protease 2; ADAM17: a disintegrin and metalloprotease 17; NRP-1: neuropilin-1; CD147: cluster of differentiation 147; GRP78: glucose regulated protein 78.
ACE2 contains a single catalytic domain with zinc-binding motif located at the extracellular side of the cell, which can be cleaved and released into the bloodstream by ADAM17 [12].
COVID-19 patients exhibit multi-organ dysfunction, due to the expression of ACE2 in the lung, heart, vascular system (endothelial cells and smooth muscle cells), brain, kidney, gut and testis [29][30][29,30]. Particularly, 80% of all ACE2-expressing cells were identified in type II pneumocyte cells, followed by the nasal and oral mucosa and alveolar macrophages [31]. ACE2 is also expressed in pericytes—undifferentiated and contractile cells that surround the capillary endothelial cells particularly in renal circulatory plexus—and in cardiac myocytes. This latter would explain the high renal microvascular damage and heart failure incidence in COVID-19 [32].
The S protein, by which SARS-CoV-2 recognizes the host ACE2 receptor, consists of three glycoprotein monomers and each monomer comprises two subunits, S1 and S2. S1 subunit can be further divided into an N-terminal domain (NTD) and a C-terminal domain (CTD). This latter domain is referred to as the ACE2 receptor-binding domain (RBD) for SARS-CoV-2 (Figure 1A) [14][16][14,16]. S1 RBD is the most variable part of the SARS-CoV-2 genome and key residue substitutions in this region can enhance the interaction ability and lead to a higher binding affinity between the S protein and ACE2 [33]. Genetic variants of SARS-CoV-2 have been emerging and circulating around the world and are frequently monitored through sequence-based surveillance, laboratory studies, and epidemiological investigations [34][35][34,35].
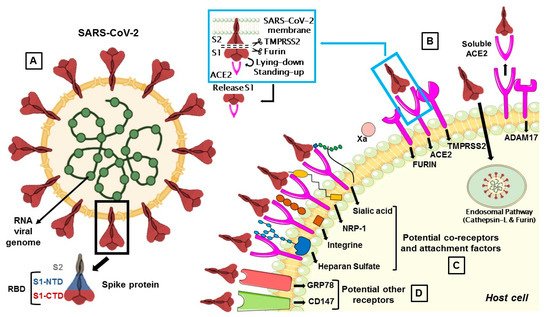
Figure 1. (A) The membrane spike protein of SARS-CoV-2 consists of three glycoprotein monomers, and each monomer contains two subunits, S1 and S2. The S1 subunit is divided into the N-terminal (NTD) and C-terminal (CTD) domain, which is referred to as the receptor-binding domain (RBD), while the S2 subunit contains membrane fusion activity. (B) SARS-CoV-2 entry mediated by TMPRSS2 and cathepsin-L. Once the S protein interacts with ACE2, TMPRSS2 cleaves S protein at the S1/S2 cleavage site and then the furin cleaves the S2 region (S2’) to initiate conformational changes for membrane fusion. The S protein can also activate ADAM17 which can cleave ACE2, resulting in shedding and soluble ACE2. In a second route, the S protein is cleaved and activated by the cathepsin-L pathway, where virions are taken up into endosomes. (C) Co-receptors are involved in SARS-CoV-2 attack. S proteins can recognize sialic acids, NRP-1, integrins and heparan sulfate for the first step of attachment to host cells. (D) CD147 and GRP78 have been identified as other potential receptors for SARS-CoV-2 entry.
There are two proteolytic activation events associated with the virus–cell membrane fusion process, mediated by the S protein, which can involve several proteases [14][15][36][14,15,36].
The first is a priming cleavage at the interface of the S1 and S2 subunit, which allows both to expose hidden cleavage sites and to increase binding affinity for the receptor. The second is a triggering cleavage that occurs within the S2 region (S2′) and allows the liberation and the conformational changes of S2 fusion peptide that mediate the fusion of the viral envelope with the host cell membrane [37].
The protease activity of ACE2 has no role in facilitating viral entry and it merely appears to act as a receptor to guide attachment and fusion of the S protein. In fact, following the binding between S1 subunit and ACE2 receptor, different serine proteases, such as TMPRSS2, cathepsin-L, and the metalloproteinase ADAM17, trigger the cleavage of ACE2 and the consequent fusion of the viral membrane to the host. Differently from SARS-CoV, the S protein of SARS-CoV-2 is activated by the serine protease furin, which facilitates conformational changes required for S1 RBD exposure and binding to surface receptors (Figure 1B) [38].
S protein has a closed and an open conformation: in the closed form, all three RBDs of the S trimer are inaccessible to ACE2 binding, while in the opened form, RBDs are exposed to ACE2 binding. After furin cleavage between the S1 and S2 domains (priming), the proportion of the S trimers in the open conformation increases. The final link between RBD and ACE2 leads to a fully open form, in which the S1 subunit remains limitedly bound to the core S2 trimer through intermediate S1 subdomains. This arrangement leaves the top of the S2 fully exposed for the next fusion of the membranes [14].
2.1. Furin Priming
Furin is a Ca2+-dependent endopeptidase, responsible for activating precursor proteins like fusion proteins of viruses. Furin is capable of cleaving and activating viral fusion proteins of HIV, Ebola [39], MERS-CoV [40], SARS-CoV [15], and SARS-CoV-2) [16] (Table 1). Furin accumulates mainly in the Golgi complex, but it can be transported to the cell surface via the endosomal pathway or can be released into the extracellular space [41]. For this reason, furin can cleave the S protein in the Golgi complex, and also in the extracellular space [38].
The addition of a furin cleavage site to S1/S2 is essential for efficient viral entry into human lung cells, and its presence expands the versatility, transmissibility and tropism of SARS-CoV-2 due to the wide cellular expression of furin proteases [42][43][42,43].
Many health conditions, like diabetes, obesity and hypertension, are associated with elevated furin levels, and this circumstance can explain why patients with such pre-existent comorbidities are subject to severe forms of COVID-19 [44]. High levels of furin suggest testing for potential furin inhibitors to counteract COVID-19. Some of them, such as the furin convertase inhibitor (chloromethyl ketone) and peptidyl chloromethyl ketones have already been reported for HIV [45], but not yet evaluated for SARS-CoV-2. There are also ongoing studies that use phytochemicals, like bromhexine and phyto-flavonoid luteolin, which are validated to block the S-protein cleavage activation and membrane fusion [46] (Table 2). Another potential candidate for adjuvant treatment is the trypsin inhibitor tamarind, which in addition to several beneficial effects on the reduction of inflammatory markers, could inhibit alone or in combination with other drugs the action of proteases that facilitate the SARS-CoV-2 infection [47].
Table 2. Targets and mechanism of action of repurposed anti-SARS-CoV-2 drugs.
| Targets | Potential Drugs | Mechanism of Action | Refs |
|---|---|---|---|
| Furin | ambroxol, bromhexine and luteolin tamarind, polyphenols, limonin and gedunin |
Block the S-protein cleavage activation and membrane fusion | [13][46][47][13,46,47] |
| TMPRSS2 | camostat, nafamostat, limonin, gedunin, otamixaban dabigatran and α-1-antitrypsin | Block S-protein cleavage mediated not only by TMPRSS2 but also by other proteases | [13][48][49][50][13,48,49,50] |
| ADAM17 | α-1-antitrypsin | Blocks S-protein cleavage | [50] |
| Syncytia formation | niclosamide, trifluoperazine, serotonin reuptake inhibitors, ivermectin | Suppress the activity of TMEM16F, involved in syncytia formation | [51][52][51,52] |
| Cathepsin L | chloroquine, hydroxychloroquine and 8P9R |
Interfere with the endosomial pathway, increasing pH | [53][54][55][56][53,54,55,56] |
| NRP-1 | heparin, natural products and small molecules | Block C-end rule peptide on NRP-1 | [57] |
| Heparan sulfate | heparin/HS mimetics | Preclude the interaction between HS and S protein | [58][59][58,59] |
| Integrins | ATN-161 | Inhibits the interaction between S protein and integrins | [60] |
| CD147 | azithromycin, cyclophilin A, doxycycline |
Interfere with ligands/CD147 interaction | [61][62][63][64][61,62,63,64] |
| GRP78 | epigallocatechin gallate, omoeriodictyol, isorhamnetin, and curcumin, berbamine, OSU-03012 | Interfere with ligands/GRP78 interaction | [65][66][65,66] |
| Factor Xa | heparin | Blocks S protein cleavage by Factor Xa and inhibits the coagulation | [20] |
| Cyclo-oxygenase-2 | non-steroidal anti-inflammatory drugs | Prevent inflammatory cytokine storm | [67] |
TMPRSS2: transmembrane serine protease 2; ADAM17: a disintegrin and metalloprotease 17; NRP-1: neuropilin-1; CD147: cluster of differentiation 147; GRP78: glucose-regulated protein 78.
2.2. TMPRSS2
TMPRSS2 is a multi-domain type II transmembrane serine protease, that mainly cleaves the S protein of SARS-CoV, MERS-CoV and SARS-CoV-2 to trigger fusion of viral envelope with the host cell membrane (Table 1). It is also important for the entry of the influenza virus into the host cell [1]. TMPRSS2 plays a dual role in the infection process: it proteolytically cleaves both the S protein, to trigger fusion of viral envelope with the host cell membrane, and the ACE2 tail, to increase the virion uptake, also through the cathepsin L -dependent pathway [68] (Figure 1).
TMPRSS2 and ACE2 are co-expressed in the lung, heart, gut, smooth muscle, liver, kidney, neurons, and immune cells [68][69][70][71][68,69,70,71]. In particular, they are both observed within the type II pneumocyte cells [72]. The TMPRSS2 gene is also expressed in the adult prostate, as reported in a research in which men were at higher risk for developing the disease with severe symptoms [73]. Indeed, the possible role of androgen receptors in increasing SARS-CoV-2 infection through the regulation of TMPRSS2 transcription has recently emerged, suggesting crosstalk between COVID-19 and prostate cancer, caused by the elevated expression of TMPRSS2 [74].
The exact sequence of cleavage events is not yet clear, but it appears that first furin cleaves at S1/S2 domain and then TMPRSS2 cleaves at the S2 cleavage site to produce fusion peptide and S2′ to trigger membrane fusion (Figure 1B) [16]. However, TMPRSS2 plays a dual role during the infection process. Beyond the afore-mentioned cleavage of the S protein, it proteolytically cleaves the ACE2 tail that event promotes the uptake of virions through the cathepsin L-dependent pathway [68].
Thus, there are two pathways for SARS-CoV-2 entry into the host cells either by endocytosis, which leads to the fusion of the viral membrane with the host cell membrane or via endosomal mechanism activated by cathepsins [75].
Given the crucial role of TMPRSS2 in favouring viral entry and the absence of approved therapies for treating the ongoing pandemic, initial attention has focused on drug repurposing opportunities to inhibit this protease (Table 2). TMPRSS2 is a protein that can be inhibited by camostat, nafamostat, and gabexate, clinically approved chemical agents in Japan for the treatment of pancreatitis [48]. There are currently several clinical trials listed on “https://clinicaltrials.gov/” to investigate the use of these repurposing drugs against SARS-CoV-2. Some data suggest that nafamostat and camostat have the potential to block S-protein cleavage mediated not only by TMPRSS2 but also by other proteases [49].
A more recent study has highlighted the correlation between a TMPRSS2 variant with a high number of cases and/or deaths of COVID-19 observed in different countries [76]. This epidemiological evidence strengthens the usefulness of TMPRSS2 inhibitors for COVID-19 management.
An in silico molecular docking study was recently performed targeting potential phytochemicals and drugs that prevent the entry of SARS-CoV-2 into host cells by inhibiting the proteolytic activity of furin and TMPRSS2 [13]. Among these, the drug nafamostat may be more beneficial than camostat in suppressing the activity of TMPRSS2. Among the phytochemicals, polyphenols in green tea were found also be potentially useful in suppressing the furin activity [13]. Limonin and gedunin found mainly in the citrus fruits and neem showed the highest binding energy at the active site of furin and TMPRSS2, respectively [13].
The activation of TMPRSS2 and other proteases also facilitates cell–cell fusion mediated by SARS-COV-2, leading to the formation of multinucleated cells, called syncytia, which contribute to tissue damage [77]. It has recently been reported that high concentrations of bromhexine, an expectorant and inhibitor of TMPRSS2 currently used in clinical trials against COVID-19 [78][79][78,79], unlike another expectorant, ambroxol, may favor the formation of syncytia [80]. These results suggest greater caution in the use of high-dose bromhexine until its effects on COVID-19 are fully understood.
From a screening of more than 3000 existing approved drugs, about 83 drugs were shown to be efficient for inhibition of S-protein-mediated cell fusion [51]. By focusing on effective drugs that also protect against virus replication and associated cytopathy, one of the most effective drugs was the anthelmintic drug niclosamide, suppressing the activity of TMEM16F (also known as anoctamin 6), a calcium-activated ion channel involved in syncytia formation [51]. In addition, it seems likely that, similar to niclosamide, all drugs, as trifluoperazine [81], serotonin reuptake inhibitors [82][83][82,83] and ivermectin [52] (Table 2), that inhibit TMEM16 proteins, block SARS-CoV-2 S-protein-induced syncytia.
2.3. Cathepsin L-Dependent P-Pathway
Cathepsins are cysteine proteases with a crucial role in protein catabolism in the endosomes and lysosomes. They require low pH (between 4.5–5.0) for optimal proteolytic activity of protein antigens resulting from pathogen endocytosis [84].
Cathepsin L is ubiquitously expressed in mammalian cells and seems to be important for SARS-CoV-2 entry in human cells as a result of cleaving the S2′ position and activate the fusion between virus and endosomal membrane, leading to the release of the viral genome into the host cells (Figure 1B) [85][86][85,86].
Drug repurposing efforts for SARS-CoV-2 have targeted endosomal cathepsins [87]. Some studies have suggested the use of the chloroquine, a broadly used antimalarial drug, and of its derivative hydroxychloroquine for COVID-19 treatment [53][54][55][88][53,54,55,88]. Chloroquine inhibited the SARS-CoV-2, SARS-CoV, influenza virus, Ebola and other viruses in vitro, perhaps through interference with the endocytic pathway [89]. However, until now, its clinical effectiveness is limited in COVID-19 patients and its potential cardiac side effects are the main concerns [53][54][55][88][53,54,55,88].
The cross-linking peptide, named 8P9R, can be another potential interferent with the endocytic pathway. Beyond its direct action of virus cross-linking activity, it could reduce the endosomal acidification inhibiting the viral entry [90]. The combined use of two endosomal acidification inhibitors (8P9R and chloroquine) improved the antiviral efficiency of the drug umifenovir, a S-protein–ACE2 fusion inhibitor, clinically available for prophylaxis and treatment of influenza in China and Russia, and now in Phase III and IV clinical trials in the USA [91] (Table 2). Since ACE2 and TMPRSS2 are individually or co-expressed in human cells, the approach of simultaneous inhibition of virus entry through blockage of both endosomal and surface fusion pathways may have better antiviral results.
2.4. ADAM17 and Soluble ACE2
The type-I transmembrane protease ADAM17 is expressed in many tissues, including lungs, muscles, heart, kidney, small intestine, pancreas, placenta, ovaries and testicles, and is involved in ectodomain shedding of cell membrane proteins [92].
The viral S protein can activate ADAM17 to cleave ACE2, resulting in a soluble ACE2 (sACE2) shedding and release (Figure 1B). How ADAM17 facilitates viral entry is not yet clear, but it might contribute to the fusion of viral particles with the host cell membrane [70].
The involvement of ADAM17 in the SARS-CoV-2 cell entry is still unclear, although it is involved in the RAS imbalance associated with virus infection [17].
According to Zoufaly et al. [93], the administration of recombinant sACE2 could act through two mechanisms either by binding to S protein and neutralizing the viral particles and the other by increasing the concentration of angiotensin II, thus helping to reduce multi-organ damage. In addition, Monteil et al. [94] showed that the administration of recombinant soluble ACE2 along with the antiviral remdesivir has an additive effect at sub-toxic concentrations and may improve the effect of remdesivir during SARS-CoV-2 infection.