1. Introduction
The NLRP3 inflammasome is a crucial step in innate immune responses and contributes to immune pathogenesis of several diseases including neurodegenerative, cardiac, pulmonary, gastrointestinal, and metabolic diseases. Therefore, inhibition of NLRP3 inflammasome activation is a novel therapeutic target for inflammation-related disorders. In recent years, many natural or synthetic molecules targeting NLRP3 have been evaluated for this purpose. Melatonin is one of the best candidate agents due to it being a natural and endogenous molecule. Figure 1 proposes melatonin's essential protective actions on the inflammatory pathways associated with NLRP3 inflammasome activation, which are reviewed in this entry. By virtue of these protective mechanisms, melatonin is a promising molecule for the treatment of inflammation-related disorders.
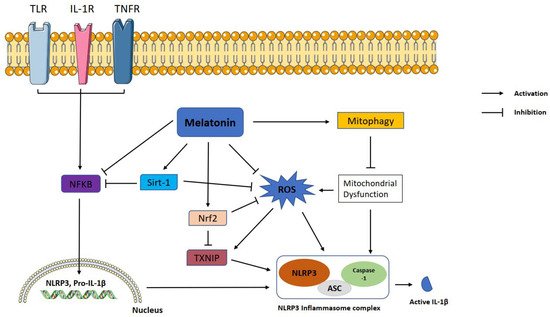
Figure 1. Representation of melatonin effects on NLRP3 inflammasome activation. Melatonin shows ameliorative effects on NLRP3 inflammasome activation via interacting with certain signaling pathways. NLRP3: NLR family pyrin domain containing 3; ASC: apoptotic-associated speck-like protein containing a caspase recruitment domain; ROS: reactive oxygen species; TXNIP: thioredoxin-interacting protein; Nrf2: nuclear factor erythroid 2-related factor 2; Sirt-1: sirtuin 1, IL-1β: interleukin 1β; NF-κB: nuclear factor kappa B.
2. Melatonin
Melatonin (
N-acetyl-5-methoxy tryptamine) is a hormone produced from L-tryptophan via a chain of enzymatic reactions, mostly in the pineal gland
[1][2][1,2]. One of the well-recognized functions of melatonin is circadian rhythm regulation. Additionally, anti-inflammatory, cytoprotective, free radical scavenging, antioxidative, anti-cancer, anti-aging, and immunomodulatory effects are also reported
[3][4][5][3,4,5]. Two different membrane receptors for melatonin binding have been identified until today: Melatonin may bind to high-affinity MT1 (Mel1a) and MT2 (Mel1b) receptors which belong to the G protein-coupled receptor (GPCR) family
[6]. Other than cell membrane receptors, melatonin can bind to the intracellular MT3 (Mel1c) receptor
[7], calmodulin
[8], calreticulin
[9], and retinoic acid receptor superfamily (retinoid Z receptors/retinoid orphan receptors (RZR/ROR)): RORα splicing variants (RORα1, RORα2, RORα3, RZRα), RZRβ, and RORγ
[10][11][10,11]. In addition to interactions with all receptors, melatonin has the ability to pass through membranes due to its amphiphilicity; so, it may react with molecules in cells without the help of receptors, termed as non-receptor mediated actions
[12]. A complete overview of anti-inflammatory actions of melatonin can be found in articles published previously
[13][14][13,14]. In this entry, we focus on the NLR family pyrin domain containing 3 (NLRP3) inflammasome, and we summarize studies on the effects of melatonin on NLRP3 inflammasome activation in a variety of diseases and possible intracellular mechanisms of these effects.
3. NLRP3 Inflammasome and Regulation
The inflammatory response is a crucial response for survival, governed by pro-inflammatory cytokines and chemokines. The innate immune system is the first to encounter pathogens and threats to the organism and respond immediately. Hyperactivation or abnormal activation of the inflammation response leads to the high concentration of cytotoxic molecules, in turn leading to tissue damage
[15]. Inflammasomes are vital contributors to innate immunity. These are cytosolic multiprotein complexes composed of a sensor protein (an AIM-like receptor or NOD-like receptor (NLR)), an adaptor protein, and an effector protein. When the inflammasomes are activated with a danger signal, cytokine secretion is triggered to clear pathogens or damaged cells
[16]. The NLR family of cytosolic pattern recognition receptors are vital in recognizing intracellular bacterial breakdown products and starting an innate immunity cascade. It has several members such as NLRP1, NLRP2, NLRP3, NLRC4, NLRP7, NLRP6, and NLRP9b
[17][18][19][20][21][22][17,18,19,20,21,22]. NLR family receptors contain a C-terminal leucine-rich repeat ligand sensing region (LRR), a NOD domain, and an N-terminal signaling module. This module may be a caspase recruitment domain (CARD), a pyrin domain (PYD), or a baculovirus inhibitor of apoptosis repeat
[16].
The most extensively studied inflammasome complex is NLRP3, and it has a vast repertoire of recognition, enabling easy activation of immunity and pathogen clearance. The complex formation is activated via internal and/or external factors; the primary factors being pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs), reactive oxygen species (ROS), ion fluxes, lysosomal destabilization, ATP, and peptide aggregates
[23]. Once activated on its LRR domain, NLRP3 recruits an adaptor protein called ASC (apoptosis-associated speck-like protein containing a CARD) on its pyrin domain. ASC oligomerizes with other recruited ASCs to form what is termed ASC specks, and in return, recruits pro-caspase-1 on its CARD containing end and triggers caspase-1 activation
[23][24][25][23,24,25]. Pro-caspase-1 is an auto-proteolytic enzyme, and the proximity induced by ASC specks thus generates caspase-1. After activation, the complex triggers inflammatory cytokine secretion which ultimately leads to pyroptosis
[26]. Canonical activation of the NLRP3 inflammasome requires two consecutive signals: priming and activation. The priming signal is initiated via TLR receptors by PAMPs such as LPS and leads to transcription of inflammatory cytokine genes through activation and nuclear translocation of NF-κB. The activation signal is the stimulation of NLRP3 by the various factors it recognizes, which eventually results in pro-caspase-1 cleavage
[23]. Cleavage of pro-caspase-1 results in two enzymatically active caspase-1 subunits: p10 and p20
[27]. Active caspase-1 cleaves pro-inflammatory cytokines IL-1β, IL-18, and the protein Gasdermin D into their mature forms. Mature Gasdermin D forms pores on the cellular membrane, resulting in pyroptotic cell death and release of cytokines
[28]. IL-1β and IL-18 amplify NLR-mediated inflammation and facilitate infiltration of immune cells
[15].
Strict regulation of NLRP3 inflammasome is crucial. Post-transcriptional, post-translational, and negative regulation mechanisms enable tightly controlled NLRP3 activation
[16]. At the post-translational level, the deubiquitinating enzyme BRCA1/BRCA2-containing complex subunit 3 (BRCC3) instigates NLRP3 activation
[29]. On the other hand, F-box L2
[30], TRIM31
[31], and MARCH7
[32] were shown to alleviate NLRP3 inflammasome activation by ubiquitination of NLRP3. Protein tyrosine phosphatase non-receptor 22
[33] and protein phosphatase 2A
[34] augment NLRP3 inflammasome by dephosphorylation, while Jun N-terminal kinase
[35] and protein kinase D
[36] augment NLRP3 inflammasome by phosphorylation of NLRP3.
Thioredoxin-interacting protein (TXNIP) regulates intracellular redox balance by inhibiting important antioxidant proteins called thioredoxins and, upon amplification of intracellular ROS levels, TXNIP dissociates from thioredoxins and induces NLRP3 activation by binding to it
[37]. Endoplasmic reticulum (ER) stress is another known inducer of NLRP3 activation: induction of ER stress and the unfolded protein response results in TXNIP upregulation
[38][39][38,39] and Ca
2+ release into the cytosol, which can damage mitochondria and lead to ROS accumulation
[40]. Mitochondrial damage can also release other stimulators of NLRP3 into the cytosol: cardiolipin and oxidized mtDNA
[41]. One more organelle that is implicated in NLRP3 activity is the lysosome: damage of lysosomes is known to release cathepsin B into the cytosol, where it induces NLRP3 activation
[42][43][42,43]. Linking these together is autophagy, which can remove damaged organelles and thus relieve the cell of inflammatory signals. Indeed, autophagy has been reported multiple times as a negative regulator of NLRP3
[44]. At the post-transcriptional level, numerous miRNAs have been demonstrated to regulate NLRP3 inflammasome; miR-223
[45], miR-7
[46], miR-1929-3p
[47] restrains NLRP3 inflammasome. Additionally, extensive studies indicate lncRNAs also have post-transcriptional regulatory effects on NLRP3 inflammasome activation; examples include nuclear enriched abundant transcript 1 (Neat1)
[48], MIAT
[49], Gm15441
[50], and Platr4
[51]. Certain molecules may bind to compounds in the NLRP3 inflammasome complex or other related molecules to inhibit it and provide negative regulation. Several proved negative regulators are B-cell adapter for phosphoinositide 3-kinase
[52], PYRIN domain-only protein 1
[53] and 2
[54], TRIM30
[55], heat shock protein 70
[56], NLR family CARD-containing 3 protein
[57] and autophagy
[58].
4. The Mechanisms of the Action of Melatonin on NLRP3 Inflammasome Inhibition
Melatonin exerts inhibitory function on NLRP3 inflammasome activation through inhibiting or activating several proteins and pathways. NF-κB is a master regulator of the priming phase of NLRP3 inflammasome activation. Melatonin prevents NLRP3 inflammasome activation by inhibiting NF-κB signaling via RORα
[59] and silent information regulator 1 (SIRT1)-dependent deacetylation of NF-κB
[59]. ROS is a main trigger of NLRP3 inflammasome activation. Growing evidence shows that melatonin reduces levels of TXNIP, leading to suppression of ROS production and NLRP3 activity
[60][61][60,61]. TXNIP is also mediator of ER stress induced NLRP3 inflammasome activation. Melatonin downregulates ER-induced TXNIP/NLRP3 pathway in LPS-induced endometritis
[61]. SIRT1 is a NAD+ dependent deacetylase and a strong regulator of inflammatory, metabolic, and oxidative stressors of cells
[62]. Preceding studies reported that sirtuins attenuate NLRP3 inflammasome activation by deacetylating of NLRP3 protein
[63], and melatonin increased SIRT1 activity to inhibit the NLRP3 inflammasome
[62][64][62,64]. Another pathway that melatonin may modulate is Nrf2 which is an antioxidant protein promoting ROS clean-up
[17]. It has been documented that melatonin displays protection against NLRP3 inflammasome activity through Nrf2-mediated ROS scavenging and elimination
[64][65][64,65]. It is well known that autophagy is a negative regulator for NLRP3 activation. Mitophagy is a subtype of autophagy that helps elimination of dysfunctional mitochondria. Melatonin increased the expression of LC3-II/LC3-I and Atg 5 as autophagy markers and Parkin and PINK-1 as mitophagy markers while suppressing NLRP3 inflammasome in the Subarachnoid hemorrhage (SAH) model
[59]. Since 3-MA, an autophagy inhibitor, reverses these beneficial effects of melatonin on NLRP3 inflammasome, the results suggest that the effect of melatonin on NLRP3 inflammasome inhibition is dependent on mitophagy induction. The regulatory function of melatonin on inflammasome partly occurs through post-transcriptional mechanisms. Melatonin inhibits NLRP3 inflammasome complex formation by altering the expression of miRNAs and long noncoding RNAs
[66][67][68][66,67,68]. Melatonin was tested against pyroptotic cell death in endothelial cells and, it reduced pyroptotic cell death via noncoding RNA MEG3/miR-223/NLRP3
[69]. Furthermore, melatonin mitigated cardiac fibrosis in mice by blocking lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome activation
[66]. Melatonin healed radiation induced lung injury both in vivo and in vitro by suppressing miR-30e/NLRP3 axis
[68].
The NLRP3 inflammasome is known to have a critical role in the pathogenesis of many diseases and conditions with inflammatory components
[70][71][72][73][70,71,72,73]; and the suppressive effect of melatonin on the NLRP3 inflammasome has been demonstrated in various in vitro and in vivo models of diseases and injuries. The detailed review of melatonin's reported effects on NLRP3 in these diseases can be found in the
full review article associated with this entry.