Adrenergic receptors are G protein-coupled receptors for epinephrine and norepinephrine. They are targets of many drugs for various conditions, including treatment of hypertension, hypotension, and asthma. Adrenergic receptors are intensively studied in structural biology, displayed for binding poses of different types of ligands.
- adrenergic receptor
- adrenoreceptor
- aminergic receptor
- GPCR
- activation mechanism
- receptor–ligand interactions
- selectivity
- allosteric modulator
- structure-based drug design
1. Introduction
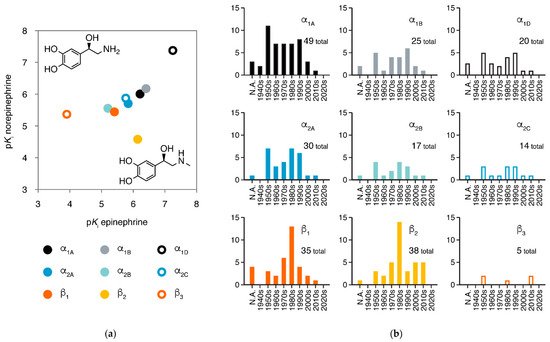
Adrenergic receptors (adrenoreceptors, ARs) are membrane proteins mediating the actions of epinephrine and norepinephrine. There are nine members of adrenergic receptors in humans (Figure 1a), widely distributed throughout the body and playing key roles in many important physiological processes, such as response to stress, control of heart rate and blood pressure, and regulation of metabolism [1][2][3][1,2,3]. Besides epinephrine and norepinephrine as emergency medicines, adrenergic receptors are also the target of many medications in treatment of various conditions, including hypertension, hypotension, heart failure, arrhythmias, and asthma [1][4][1,4]. In the 21st century, the development of new drugs against adrenergic receptors has slowed down, but the once most-neglected member β3-adrenergic receptor is raising more attention than ever (Figure 1b).
The β2-adrenergic receptor (β2AR) is a prototype GPCR for studies of sequence, structure, and function: β2AR is the first GPCR cloned [11][5], the second GPCR was determined for atomic structure [12][13][6,7] (only after rhodopsin), and the first GPCR solved for the active state structure in a functional complex [14][8] (with Gsprotein heterotrimer). β-arrestins, the cytoplasmic partners responsible for desensitization of most GPCRs, were named after the β2AR [15][16][9,10]. Turkey β1AR has been comprehensively studied in structural biology (in this article, all structure of β1AR refers to turkey β1AR unless noted).
β2AR and β1AR are among the GPCRs with the largest numbers of structures obtained, providing rich information for ligand recognition and activation mechanisms (β2AR has 38 structures for 19 ligands and β1AR has 30 structures for 17 ligands). In 2019, structures of the three α2adrenergic receptors were also reported, disclosing the structural basis for the receptor-type selectivity of ligands. In this review, we summarized current achievements of structural study in adrenergic receptors and advances in drug discovery based on these structures.
2. Mechanisms of β Adrenergic Receptors
About 90 receptors [17][52] share a common mechanism: the conformational changes are mediated by the microswitch of the P5.50-I3.40-F6.44 motif, induced by an agonist. How agonists trigger the microswitch of the P-I-F motif largely varies in different receptor families. For β2AR or β1AR, an agonist forms a hydrogen bond to S5.46, thus pulling TM5 closer to TM3/6. An antagonist, in contrast, lacks this hydrogen bond and tends to occupy the space with hydrophobic moiety, thus preventing the movement of TM5 (Figure 2b).
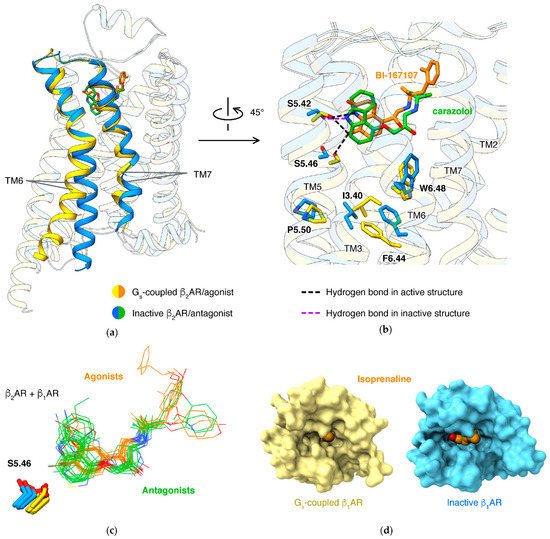
Binding of the orthosteric agonist and coupling of G protein have an effect on each other: agonists have higher affinities in β2AR with the presence of G protein than in the absence of G protein [34][53]. Structure studies of agonists binding in different states of β1AR disclosed the molecular basis: active state structures have smaller pockets when comparing to agonist-bound inactive state structures (Figure 2d) [21][54]. This was established in different types of class A GPCRs, including lipid receptors [35][36][37][38][55,56,57,58], peptide receptors [39][40][41][59,60,61], and nucleotide receptors [42][43][44][45][62,63,64,65].
For βARs, separately targeting β-arrestin pathways or G protein pathways is considered a therapeutic potential for diseases, including heart failure and asthma [46][47][78,79]. Correspondingly, β1AR forms interactions to arrestin with more residues in TM2/7 and fewer residues in TM5/6 of19F-labeling on the cytoplasmic ends of TM6 and TM7 showed that arrestin-biased agonists induce more conformational change of TM7 than of TM6 for β2AR in solution [48][84]. Structures showed that arrestin-biased agonists carvedilol [49][85], bucindolo [50][86], formoterol [51][87], and BI-167107 [52][88] have an aryl-substituted alkyl tail attached to the amino N atom, and the bulky aromatic rings occupy an extended pocket formed by TM2/3/7 and ECL2 in β2AR
NMR [48][53][54][84,93,94], single-molecule fluorescence resonance energy transfer [55][95], and double electron–electron resonance [54][56][94,96] results showed that partial agonists induce a smaller shift of conformational equilibrium of TM6, compared to full agonists. A well-identified key interaction affecting ligand efficacy (ratio of maximal response of the ligand to full response) is a hydrogen bond to N6.55 by a full agonist, but not a partial agonist. Mutations of N6.55 and S5.43 (which forms hydrogen bond to N6.55) in β2AR reduce the efficacy of full agonist isoproterenol, strongly supporting that this hydrogen bond network is crucial to the full activation of βARs [26][70]. All full agonists can form a hydrogen bond to N6.55, either by 3-hydroxy in catechol (like epinephrine [19][20][31,43] or isoproterenol [21][22][54,66]) or groups mimicking catechol (such as formoterol [18][11] or BI-167107 [14][20][57][8,43,90]).
β1AR and β2AR are highly similar in sequence (identity 57% at full-length and 70% at pocket residues) and structure (root mean square deviation 0.71 Å for inactive state and 1.00 Å for Gs-coupled active state). Despite their high homology, numerous agonists and antagonists selective for β1AR or β2AR have been discovered. Even endogenous agonist norepinephrine, small in size, thus all the interacting residues are the same in the two receptors, is approximately 10-fold selective for β1AR (while the other endogenous ligand epinephrine, with only one extra methyl group, is nonselective).
In β1AR and β2AR, although all the residues at the orthosteric site are the same, some residues at the edge of the pocket are different. Large ligands may directly interact with these residues. Therefore, ligands potentially take different paths to enter β1AR and β2AR. This hypothesis explains the fast association rate of norepinephrine in β1AR and is supported by free energy profiles, calculated using metadynamics simulations [20][43].
3. Allosteric Modulations of β2 Adrenergic Receptor
In a GPCR, a ligand may bind outside the orthosteric site and stabilize a specific state, and this phenomenon is called allosteric modulation [58][59][105,106]. An allosteric ligand increasing the signaling of orthosteric agonists is a positive allosteric modulator (PAM), and one decreasing the signaling is a negative allosteric modulator (NAM). The most effective way to identify an allosteric site is determining the structure of the GPCR in complex with the allosteric modulator [60][107]. Structures of β2AR captured small molecules binding at different sites and exhibited how they affect the signaling (Figure 3).
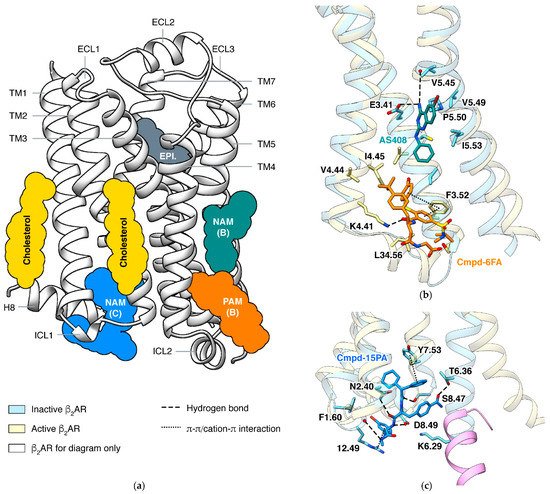
In β2AR, both PAM and NAM binds in this groove are identified by crystal structures (Figure 3b). The PAM and the NAM locate at different heights and stabilize different states through specific interactions: the PAM Cmpd-6FA binds close to the cytoplasmic end and interacts with ICL2 and adjacent residues on TM3 and TM4 [62][110]. Since functions of ICL2 and TM5 are common in activation process of class A GPCRs, it is not surprising that allosteric modulators binding at the same site were also discovered in other receptors: PAMs AP8 [63][112] and Cpd-1 [64][113] of free fatty acid receptor 1, PAM LY3154207 [65][114] of dopamine receptor D1, and NAMs
GPCRs use the cytoplasmic surface to interact with intracellular partners, including G proteins and β-arrestins. Small molecules binding at this site were mainly discovered in chemokine receptors [66][67][68][69][116,117,118,119], while Cmpd-15PA in β2AR [70][120] is the only case outside the chemokine Cmpd-15PA has little collisions with G protein but stabilizes the inactive state of the receptor through extensive interactions with TM1, TM2, TM6, TM7, H8, and ICL1. Although β2AR and β1AR only differ in one position (F1.60 in β2AR and T1.60 in β1AR) among all residues interacting with Cmpd-15PA, the Cmpd-15 (parent compound of Cmpd-15PA with comparable effects) decreases the EC50of orthosteric agonist in only β2AR
4. Structure-Based Ligand Discovery of Adrenergic Receptors
Structure-based virtual screening can identify ligands with novel scaffolds and chemical types [71][72][122,123]. Since structures of β2AR are reported, new antagonists and agonists were discovered through virtual screening with molecular docking. Docking screening identified a new antagonist with benzhydrylpiperazine moiety having high sub-nM affinity (Ki= 0.311 nM) [73][124].
Structures can guide modification of known ligands to achieve new function. Two designed β agonists, analogs of BI-167107 and epinephrine but that can form disulfide bond, covalently bind to β2AR with designed mutation Adding of a methyl group to cyanopindolol (partial agonist of β1AR and β2AR) at a position close to S5.46 further reduced its efficacy [33][77]. Inspired by structures of β adrenergic receptors, five compounds based on atypical antipsychotic drug aripiprazole were designed and shown to have different effect on dopamine receptor D2(one unbiased agonist, two arrestin-biased agonists, and two antagonists) [74][129].
Molecular docking aims to identify compounds with high affinity but does not distinguish between an agonist and antagonist. Molecular interaction fingerprints derived from structures of a GPCR with various ligands have been developed to predict the actions of ligand. When only the first inactive β2AR structure was available, the molecular interaction fingerprint was generated based on docking poses of ligands [75][130]. The newest version of fingerprint was developed based on 31 structures of β1AR
Structure-based drug design against GPCRs relies on a large number of solved structures binding various ligands. Since 2018, many structures of active state GPCRs in functional complexes have been solved with the rapidly developed cryogenic electron microscopy (cryo-EM) technique. A remarkable case is dopamine receptor D1: no structure was reported before February 2021, and then 11 structures were released within two months [64][76][77][78][113,133,134,135]. For adrenergic receptors, up to April 2021, a total of eight cryo-EM structures were published: one is arrestin-coupled β1AR and the others are G protein-coupled (four β2AR, Gs[14][24][25][8,68,69]; one β1AR, Gs[22][66]; two α2BAR, Giand Go[17][52]).
Crystallography is still powerful for the structural study of adrenergic receptors. β1AR can co-crystallize with small, fragment-like ligands [79][89]. For β2AR, structures in complex with eight ligands were obtained using a newly developed transient ligand exchange method termed Complex-LCP (Crystallization of Membrane Proteins using transient Ligand EXchange in LCP). The Complex-LCP method relies on the serial femtosecond crystallography (SFX) data-collection approach, using extremely bright and short pulses generated by an X-ray free-electron laser (XFEL), thus collects data from small crystals at room temperature [31][75].