Urine is perhaps, of all biofluids, the one with greater potential in clinical peptidomics. Urinary proteins and peptides originate from the secretions of renal tubular epithelial cells, shedding of cells along the urinary tract, exosome secretion, and more importantly, from glomerular filtration of plasma. Therefore, beyond the renal system’s pathophysiological status, the urinary peptidome is influenced by systemic disturbances.
- urine
- peptides
- proteases
- peptidomics
- degradomics
- biomarkers
- predictive
- preventive and personalized (3P) medicine
- molecular patterns
- individualized patient profiling
1. Introduction
The peptidome refers to the whole set of low molecular weight (LMW) One of the key features of peptidomics, i.e., the analysis of the native peptidome, is studying endogenous proteolytic events while preserving information on post-translational modifications [4][1]. The human degradome comprises over 550 proteases, categorized into five groups, according to the catalytic mechanism [6][2]: serine proteases, threonine proteases, aspartic proteases, cysteine proteases, and metalloproteases. Many disease states are associated with an unrestricted and abnormal progression of proteolysis [7][3], making proteases and peptides key molecular entities with value as biomarkers and potential therapeutic targets.
There are many advantages of peptidomics over proteomics: (1) it can attain a larger number of disease-specific analytes, provided the generation of many peptides from a single parent protein, which results in improved discrimination; (2) it allows the study of the disease microenvironment, as peptides can cross endothelial barriers providing clinically-relevant biomarkers; (3) it is possible to extract information associated to proteolytic activity; (4) sample processing is much simpler, excusing enzymatic digestion and reducing intra-sample heterogeneity [9,10][4][5].
Endogenous peptides have only recently received attention as their relevance in disease characterization has not been adequately acknowledged [10][5]. However, today, the combination of high-performance liquid chromatography or capillary electrophoresis with high resolution mass spectrometers, usually using time-of-flight analyzers, but also Orbitraps, provides the needed sensitivity for the identification of thousands of peptides. Furthermore, the detection of LMW protein species might be hampered by the presence of highly abundant proteins in some biofluids, such as blood, thus requiring pre-fractionation or enrichment steps [13][6]. Some endogenous peptides can also be shared over several proteins, obfuscating their role in disease etiology [14][7].
Urine is perhaps, of all biofluids, the one with greater potential in clinical peptidomics. Urinary proteins and peptides originate from the secretions of renal tubular epithelial cells, shedding of cells along the urinary tract, exosome secretion, and more importantly, from glomerular filtration of plasma [15,16][8][9]. Thus, peptides dominate a relatively larger proportion of the proteome in urine as opposed to other biofluids. Finally, the urine peptidome is considered more stable than other peptidomes, such as blood, because the bulk of proteolytic events has already been completed within the bladder [5][10].
The particular clinical value of urine peptidome is reflected by several studies where putative biomarker panels have been developed based on urinary peptides, aiming at the non-invasive diagnosis, prognosis of specific diseases, or flagging the need of an intervention. For instance, peptidomics has been used for the investigation of renal system diseases such as for the prediction of chronic kidney disease (CKD) progression or remission [16[9][11],18], suggesting associations of multiple peptides, predominantly collagen fragments, with glomerular filtration and the processes of inflammation and repair [19][12]. Another example is the so-called CKD273 classifier, a panel composed of 273 urinary peptides that has been validated in multiple studies for early detection of CKD and monitoring progression [18,20][11][13] and even recently used in a clinical trial for diabetic patient stratification and early detection of diabetic nephropathy [21][14]. (2016) also studied peptidome alterations related to Autosomal dominant polycystic kidney disease (ADPKD) and acute kidney injury (AKI) and proposed a biomarker panel of 20 LMW urine peptides for ADPKD and 39 for AKI [22][15].
Apart from the proven value of urine peptidomics in developing new diagnostic tools for diseases afflicting the renal-urogenital axis, the diagnostic/prognostic potential of urine peptidomics extends to other systemic conditions. Endogenous peptides from other sources may cross the endothelial barrier and be cleared out by the renal system, thus being surveyed through urine analysis [16,32,33][9][16][17]. Besides, a 96-peptide classifier—the heart failure predictor—was proposed to stratify the risk for left ventricular heart failure [36][18]. On top of peptides, proteases predicted and validated from urine peptidome may improve our perception of the disease and contribute with new insight and tools for the diagnosis/prognosis.
Clinical interest in urine peptides is mounting, particularly as surrogate biomarkers for chronic conditions, such as those directly afflicting the kidney. With the growing number of studies following a urine peptidomics approach, it is mandatory to evaluate the data published thus far to typify the major findings in this field, aiming to accelerate the introduction of urine peptides as biomarkers in clinical practice. Furthermore, by exploring peptides’ physical properties, such as mass, length, or isoelectric point, we hope to disclose disease class patterns that may be of diagnosis relevance upon a simple urinalysis. Finally, considering the urine’s unique proteolytic environment, which deems the urine peptide collections more stable than in any other biofluid, we might predict potentially dysregulated proteases in the various conditions, hopefully pinpointing new biomarkers to be added to the set of urinary peptides.
2. Brief Methods
A literature data research was conducted using the PubMed database up to July 2020, employing the combination of the following keywords: (“peptidome” OR “peptidomic” OR “peptidomics”) AND (“urine” OR “urinary”). This search retrieved 132 articles. After excluding reviews, articles written in languages other than English and not directly related to the urinary peptidome, and after including some articles found by cross-referencing the identified papers, a final set of 54 was obtained. Not surprisingly, studies on urinary peptidome in renal diseases represent the largest fraction (~36%), closely followed by those on cardiovascular abnormalities (~20%).
Each study was mined to extract the pathology, disease class, general information about the discovery and validation phases (e.g., number of subjects, number of identified proteins and peptides, number of sequenced differentially expressed peptides, and number of sequenced signature peptides), methodological approach (identification strategy, sequencing strategy, and biomarker assessment algorithm) and, if present, the details on the peptide panel performance in terms of C-statistics. Then, to probe the hypothetical diagnostic nature of the peptides across all conditions/disease classes, the peptides displaying a potential discrimination power amongst conditions were selected from the discovery phase or from the validation phase, for instance, in the cases where the former were not available.
All peptide sequences associated with a disease were given a unique peptide ID and were associated to a specific condition, classified in nine major classes: autoimmune, bowel, cancer, cardiovascular, infection, mental, metabolic, renal, and respiratory diseases. Similarly, all peptides identified in the Di Meo et al. [38][19] dataset were given a unique peptide ID and associated to a single class: health.
Aiming to identify peptide signatures for each condition, we performed network analysis using Cytoscape (v.3.8.2). The conditions were defined as source nodes and the sequences as target nodes. We then mapped the nodes according to the degree, i.e., the total number of interactions, to single out those peptides associated with a unique condition (degree = 1). The node size was defined in descending order of the degree (with a bypass for the conditions themselves) to highlight those peptides with higher potential to create a disease fingerprint.
The signature peptides obtained through network analysis were then studied regarding their single amino acid composition and physical-chemical properties, using R statistical software built-in functions and the package ‘Peptides’ [39][20]. As a reference, we used the complete set of peptides identified in healthy individuals [38][19]. First, we performed contingency analysis of the amino acid composition. prediction tool available onhttp://www.camp3.bicnirrh.res.in/, accessed on 23 July 2020) and the content in proline, an amino acid with special properties.
Provided the possibility to study the urine degradome, based on its peptidome, we used Proteasix web tool, an open-source peptide-centric tool, to compute the cleavage probability of native proteins originating the urinary peptides by a wide array of proteases [40][21]. In this analysis, both N-terminal and C-terminal cleavages were considered, but exclusively by human proteases. Proteasix predicts proteases based on the known target sequence specificity, according to the peptidase database MEROPS [41][22]. The list of predicted proteases was then imported to R and the enrichment over the health dataset was calculated.
3. Results and Discussion
Although we only included one study unequivocally reporting the healthy urine peptidome, the number of sequenced peptides is comparable to the sum of disease peptides: 3765 unique peptides were gathered to represent the healthy urinary peptidome [38][19]. A quick comparison between disease and health proteome and peptidome shows that while 30% of the proteins (88 out of 294) dysregulated in disease were present in the comprehensive health dataset (Table S4), only 17.4% of the dysregulated of the peptides (203 out of 1166) were also sequenced in the healthy patients. This supports the notion that biomarkers panels composed of peptides can more easily discriminate a disease than a protein panel. Nonetheless, this first analysis: (i) corroborates the vision of urine as an important reservoir of disease biomarkers; (ii) demonstrates the superiority of urine peptidomics to proteomics in biomarker discovery and (iii) suggests that urine’s proteolytic environment underlies the greater diversity of surrogate peptide markers compared to protein markers.
Aiming to uncover and identify peptides with biomarker ability for the 37 conditions studied (spread across nine major classes), a network analysis was performed using Cytoscape. The conditions are presented as red nodes (source) and the peptide sequences as blue nodes (target). As shown by the density of larger blue nodes in the network’s outer layer, there are many peptides with the potential of becoming a signature for many of these diseases–644 in total. Beyond being a reservoir of molecular markers for kidney conditions, such as chronic kidney disease (with 43 specific peptides) or type 2 diabetic nephropathy (with 54 specific peptides), this exploratory analysis illustrates that urine is an important source of putative biomarkers for diseases afflicting extra-nephrotic tissues/organs, namely the heart (e.g., heart failure with reduced ejection fraction with 11 specific peptides), the lungs (e.g., chronic obstructive pulmonary disease with 38 specific peptides) or even the bowel (e.g., necrotizing enterocolitis with eight specific peptides).
Being the most relevant disease-specific urine peptides probed to date, not only regarding health but also considering all other conditions whose the urine peptidome has been investigated, we explored their physical properties in different disease classes hoping to disclose distinctive molecular traits among them. Due to the low representation of some classes in terms of peptides, we only considered the following classes: autoimmune, cancer, cardiovascular, renal, and respiratory diseases. Considering that the amino acids confer the first level of complexity of peptides and are the first determinants of the physical, chemical, and biological properties of peptides, it is not implausible to hypothesize that the amino acid composition, alone, might distinguish different disease classes. Interestingly, autoimmune peptides are characterized by the lack of Cys and Met.
More evident is, perhaps, the higher content in Gly of the disease-associated peptides. A new hypothesis emerges, whereby, the relative increase of Gly on urine peptidome indicts the presence of a disease. Also noticeable is the higher levels of Pro in peptides more specific to cardiovascular and respiratory conditions, suggesting that this amino acid might be a nonspecific marker of these disease classes. [44][23] have already described the arginine and proline metabolism dysregulation in a leporine model of atherosclerosis after characterizing the urine metabolome.
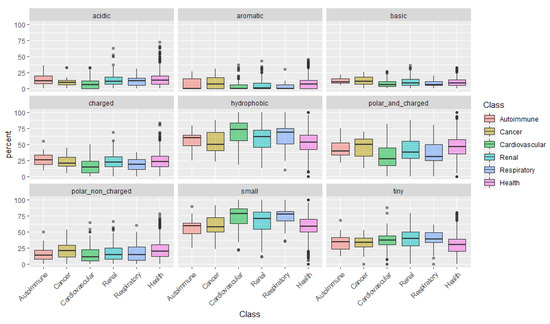
After examining the amino acids individually, we categorize them according to their physical-chemical properties: acidic, aromatic, basic, charged, hydrophobic, polar and charged, polar non-charged, small, and tiny amino acids (Figure 31). This analysis uncovered a pattern in peptides associated with cardiovascular conditions: peptides are mainly composed of small and more hydrophobic amino acids, with low representation of polar and/or charged amino acids.
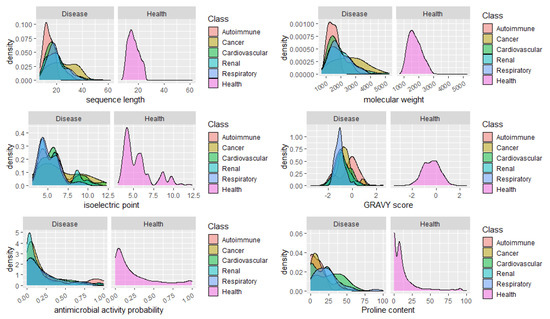
Finally, we analyzed the distribution of peptides in health and disease sets concerning relevant features and properties of the peptides, such as the sequence length, molecular weight, isoelectric point, GRAVY score, and the content in Proline. It would be expected a high percentage of peptides with predicted antimicrobial peptides in urine from patients with infections, however we did not include the peptides associated to infections in this analysis, given the low number of specific peptides found (14). No apparent major changes in the distribution of the peptides regarding the isoelectric point could be seen, as well. Concerning the GRAVY score, a commonly used metric to evaluate a peptide’s hydrophobicity, a very sharp distribution of peptides around -1
Could the biological foundation of this observation reside on the operation of more stringent proteases, resulting in lesser cleavage events and, thus, in, averagely longer and heavier peptides? Particularly, it would be important to assess if, for instance, in cancer risk models, the addition of the average peptide length or mass on top of clinical variables could improve the model’s predictive power. Finally, the distribution of peptides according to the content in Pro, evidenced once more, the predominance of Pro-rich peptides in cardiovascular diseases. See, for instance, the large proportion of peptides composed of 40–60% Pro in cardiovascular conditions in Figure 42.
Glancing over the diversity of urinary peptides associated with unrelated pathologies, we hypothesized that predicting the most active proteases would give another layer of discrimination between such conditions. From over 550 proteases composing the human degradome, we predicted the activity of 74 proteases, from four different catalytic types: aspartic (4), cysteine (13), metalloproteases (22), and serine (35) proteases, acting on either the N- or C-termini of the urine proteins/peptides. We started by looking at the proteases which are more susceptible to dysregulation in disease. For such purpose, we computed the fold-enrichment over the proteases predicted to be active in health.
As depicted in Figure S1, we could not calculate the fold-enrichment for two serine proteases from the complete set of predicted proteases as these were only predicted in disease. Following metalloproteases, serine proteases are the second class more prone to dysregulation, noticeably tripeptidyl-peptidase 1 (TPP1), a lysosomal protease acting mainly on hydrophobic proteins. Of note, only three proteases were predicted exclusively from the healthy peptidome: beta-secretase 1 (BACE1), an aspartic protease responsible for the proteolytic processing of amyloid precursor protein, plasma kallikrein (KLKB1), a serine protease with a key role in hemostasis, and trypsin-3 (PRSS3), a serine protease involved in digestion, among other processes. Whether their absence in the disease urine degradome is due to the increased expression of protease inhibitors remains to confirm.
To investigate this more in-depth, we used a similar strategy to gene ontology enrichment analysis to identify overrepresented proteases compared to health status. To identify which proteases were significantly enriched across diseases/classes, we performed a hypergeometric test and corrected for multiple testing by the Bonferroni method. For instance, the aspartic protease gastricsin (PGC) is significantly enriched exclusively in autoimmune diseases. Serine proteases, remarkably hepsin (HPN), suppressor of tumorigenicity 14 (ST14), and transmembrane protease serine 6 (TMPRSS6) are significantly enriched in bowel diseases.
This analysis shows that the protease profile increases our insight regarding differences between several conditions, showing exciting clinical potential. For instance, once again, MMP10 stands out as an overactive protease in many conditions, such as renal, metabolic, and cardiovascular diseases. This heatmap also highlights the activation of aspartic proteases in cancer. Curiously, an end-stage renal disease in the setting of autosomal dominant polycystic kidney disease is mainly characterized by the activation of caspases.
Aiming at inspecting the specificity of the putative proteases to every disease at scope, we built a map with Cytoscape showing all significant associations as deemed by the hypergeometric test (the reader is referred tohttps://public.ndexbio.org/#/network/8c07b5f7-a8e3-11eb-9e72-0ac135e8bacf?accesskey=653ffb5202553f1d3db97c4aeac15c0b1500efdf67e70f85a6d7b5478c3901c5) for an interactive exploration of each condition’s putative degradome signature). Proteases (outer circle in the network) are arranged in a counterclockwise ascending spiral, highlighting an increasing specificity. Due to proteases’ participation in many biological processes that are common to many pathological conditions, it is not surprising to verify that only seven out of 37 conditions included in this study can, theoretically, be identified by higher activity of a single protease. This comes with no surprise, given the association of diabetes with a hypercoagulable state of the blood and the fact that this protease has been pointed as a novel therapeutic target for this condition [46][24].
This may be explained by lymphocytes’ activity, which releases granzymes to induce apoptosis of virus-infected cells, often resulting in collateral damage to noninfected tubule cells [47][25]. It explains the activation of both the intrinsic and extrinsic pathways of apoptosis, a major hallmark of this disease [48][26]. The protease granzyme B (GZMB, 3.3-fold enrichment), which is also known for its role in stimulating apoptosis, was predicted exclusively from peptides associated with necrotizing enterocolitis, an association that has not yet been reported. According to our analysis, acute rejection of kidney transplant can also be monitored by assessing the activity of the protease disintegrin and metalloproteinase domain-containing protein 17
Apart from these seven exceptions, we could sketch a minimal degradome fingerprint for many (21) of the remaining conditions (30), following the same rationale applied to peptides, where often these are integrated into multiplex panels to improve the accuracy of the diagnostic test. In Table 1, one can find the minimal combination of predicted proteases that identify a given disease, signatures which deserve further consideration in future biomarker studies. Even for the eight conditions that failed to show a specific degradome, the combination with specific peptides may be the key to develop a sensitive and specific diagnostic panel. Regardless of the nature of the biomolecules, peptides or proteases herein identified or predicted should, in principle, foster new avenues of research towards biomarker implementation in the era of 3PM [51][27].
Table 1. Putative minimal degradome signature for all conditions studied through urine peptidomics.
| Condition 1 | Class | Minimal Degradome Signature |
|---|---|---|
| Type 2 diabetic nephropathy | Renal | F10 2 |
| BK virus nephritis | Infection | GZMK or TPSAB1 2 |
| End-stage renal disease in the setting of autosomal dominant polycystic kidney disease | Renal | CASP2 or CASP8 |
| Necrotizing enterocolitis | Bowel | GZMB |
| Acute rejection of kidney transplant | Autoimmune | ADAM17 |
| Schistosoma haematobium infection | Infection | PGA3 |
| Major depressive disorder | Mental | PCSK2, HTRA2 or CELA1 |
| Acute Kawasaki disease | Cardiovascular | CAPN1 + MMP7 |
| Bladder cancer | Cancer | CTSE + MCPT3 |
| Lupus nephritis | Renal | PITRM1 + PGC |
| Renal cell cancer | Cancer | KLK3 + CTSK |
| Preeclampsia | Cardiovascular | CTSK + BMP1 |
| Diabetes mellitus | Metabolic | MMP17 + BMP1 |
| Autosomal dominant polycystic kidney disease | Renal | KLK6 + MMP9 |
| Left ventricular diastolic dysfunction and hypertension | Cardiovascular | BMP1 + TMPRSS11D |
| Prostate cancer | Cancer | CTSE + MMP2 |
| Helicobacter pylori infection | Infection | ADAMTS4 + KLK4 |
| Diabetic nephropathy versus chronic renal disease | Renal | GZMA + MMP10 |
| Acute kidney injury | Renal | (CASP3 or CASP6) + (ADAMTS4 or MMP2) |
| Anti-neutrophil cytoplasmic antibody-associated vasculitis | Autoimmune | MMP17 + CTSD + PGC |
| Type 2 diabetes mellitus | Metabolic | CAPN1 + CAPN2 + ELANE |
| Type 1 diabetes mellitus | Metabolic | CTSK + ADAM10 + CASP1 |
| Systemic juvenile idiopathic arthritis | Autoimmune | ADAM10 + CASP1 + F2 |
| Left ventricular diastolic dysfunction | Cardiovascular | MMP9 + TMPRSS11D + THOP1 |
| Chronic kidney disease | Renal | ADAMTS4 + MMP9 + MMP25 |
| Type 1 diabetes mellitus versus Type 2 diabetes mellitus | Metabolic | KLK14 + KLK2 + MMP3 |
| Chronic obstructive pulmonary disease with alpha-1-antitrypsin deficiency | Respiratory | TMPRS11D + KLK6 + NLN |
| Chronic allograft nephropathy or dysfunction | Renal | GZMA + PCSK5 + (F2 or TMPRS11D) |
4. Conclusions
In the everlasting quest of finding new and more specific disease biomarkers, either in a single or in a multiplex fashion, urine peptidomics and degradomics progressively occupy a relevant position in the most fruitful methodologic approaches. Urine is simultaneously a simpler biological matrix to uncover biomarkers, with less interfering substances than blood-derived products, and a depot of substances from the entire body, virtually allowing to monitor molecular alterations occurring as a consequence of any disease. Furthermore, the peptidome, while technically simpler to analyze, is more complex than the proteome, which may increase the odds for discriminating diseases based on a combination of analytes, not to mention that peptidomics is a good source of information for the characterization of the degradome–itself another source of surrogate markers.
The analysis of all peptidome datasets available today demonstrated its great potential towards biomarker discovery, as shown by the tenths of specific peptides for various conditions afflicting different systems, such as the renal, cardiovascular, pulmonary, and metabolic ones. These peptides are disclosed without reserve assupplementary materialand their diagnostic potential merit further scrutiny. Moreover, our analysis suggests that peptides physical-chemical properties may themselves help improve the robustness of disease-predicting models. Particularly, it might be interesting in the future, to test the discriminatory value of proline content for cardiovascular diseases and the value of sequence length/mass for the diagnosis of cancer.
Finally, the present study shows that peptidomics is a double source of information, whose potential extends beyond the insight on dysregulated peptides, by allowing to unveil dysregulated proteases in disease, making use of predictive tools (Proteasix). The prediction of the degradome from the most comprehensive urine peptidome dataset reunited to date revealed a remarkable specificity of the granzymes B and K, caspases 2 and 8, pepsin A-3 and of the disintegrin and metalloproteinase domain-containing protein 17, neuroendocrine convertase 2, serine protease HTRA2 (mitochondrial) and the chymotrypsin-like elastase family member 1 which were associated with a single condition. Ultimately, this study advocates the combination of a urine peptidomics-degradomics approach for the discovery and development of new biomarker panels regardless of the origin of the disease. These molecular patterns can potentially be used in any of the three main axes of 3PM, from predicting to preventing and personalizing medical treatment, with the great advantage of a noninvasive monitoring of the disease evolution or remission through a liquid biopsy.
References
- Sarkizova, S.; Klaeger, S.; Le, P.M.; Li, L.W.; Oliveira, G.; Keshishian, H.; Hartigan, C.R.; Zhang, W.; Braun, D.A.; Ligon, K.L.; et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020, 38, 199–209.
- Puente, X.S.; Sánchez, L.M.; Overall, C.M.; López-Otín, C. Human and mouse proteases: A comparative genomic approach. Nat. Rev. Genet. 2003, 4, 544.
- Lee, P.Y.; Osman, J.; Low, T.Y.; Jamal, R. Plasma/Serum proteomics: Depletion strategies for reducing high-abundance proteins for biomarker discovery. Bioanalysis 2019, 11, 1799–1812.
- Latosinska, A.; Siwy, J.; Mischak, H.; Frantzi, M. Peptidomics and proteomics based on CE-MS as a robust tool in clinical application: The past, the present, and the future. Electrophoresis 2019, 40, 2294–2308.
- Di Meo, A.; Pasic, M.D.; Yousef, G.M. Proteomics and peptidomics: Moving toward precision medicine in urological malignancies. Oncotarget 2016, 7, 52460–52474.
- Cai, T.; Yang, F. Strategies for characterization of low-abundant intact or truncated low-molecular-weight proteins from human plasma. Enzymes 2017, 42, 105–123.
- Klein, J.; Bascands, J.-L.; Mischak, H.; Schanstra, J.P. The role of urinary peptidomics in kidney disease research. Kidney Int. 2016, 89, 539–545.
- Štěpánová, S.; Kašička, V. Analysis of proteins and peptides by electromigration methods in microchips. J. Sep. Sci. 2016, 40, 228–250.
- Pontillo, C.; Mischak, H. Urinary peptide-based classifier CKD273: Towards clinical application in chronic kidney disease. Clin. Kidney J. 2017, 10, 192–201.
- Lygirou, V.; Latosinska, A.; Makridakis, M.; Mullen, W.; Delles, C.; Schanstra, J.P.; Zoidakis, J.; Pieske, B.; Mischak, H.; Vlahou, A. Plasma proteomic analysis reveals altered protein abundances in cardiovascular disease. J. Transl. Med. 2018, 16, 104.
- Markoska, A.; Valaiyapathi, R.; Thorn, C.; Dornhorst, A. Urinary C peptide creatinine ratio in pregnant women with normal glucose tolerance and type 1 diabetes: Evidence for insulin secretion. BMJ Open Diabetes Res. Care 2017, 5, e000313.
- Schanstra, J.P.; Zürbig, P.; Alkhalaf, A.; Argiles, A.; Bakker, S.J.L.; Beige, J.; Bilo, H.J.G.; Chatzikyrkou, C.; Dakna, M.; Dawson, J.; et al. Diagnosis and prediction of CKD progression by assessment of urinary peptides. J. Am. Soc. Nephrol. 2015, 26, 1999–2010.
- Pontillo, C.; Mischak, H. Urinary biomarkers to predict CKD: Is the future in multi-marker panels? Nephrol. Dial. Transplant. 2016, 31, 1373–1375.
- Tofte, N.; Lindhardt, M.; Adamova, K.; Bakker, S.J.L.; Beige, J.; Beulens, J.W.J.; Birkenfeld, A.L.; Currie, G.; Delles, C.; Dimos, I.; et al. Early detection of diabetic kidney disease by urinary proteomics and subsequent intervention with spironolactone to delay progression (PRIORITY): A prospective observational study and embedded randomised placebo-controlled trial. Lancet Diabetes Endocrinol. 2020, 8, 301–312.
- Markoska, K.; Pejchinovski, M.; Pontillo, C.; Zürbig, P.; Dakna, M.; Masin-Spasovska, J.; Stojceva-Taneva, O.; Mischak, H.; Spasovski, G. MO024 urinary peptide biomarkers associated with improvement in eGFR in CKD patients. Nephrol. Dial. Transplant. 2016, 31, i38.
- Magalhães, P.; Pontillo, C.; Pejchinovski, M.; Siwy, J.; Krochmal, M.; Makridakis, M.; Carrick, E.; Klein, J.; Mullen, W.; Jankowski, J.; et al. Comparison of urine and plasma peptidome indicates selectivity in renal peptide handling. Proteom. Clin. Appl. 2018, 12, 1700163.
- Sirolli, V.; Pieroni, L.; Di Liberato, L.; Urbani, A.; Bonomini, M. Urinary peptidomic biomarkers in kidney diseases. Int. J. Mol. Sci. 2019, 21, 96.
- Zhang, Z.-Y.; Ravassa, S.; Nkuipou-Kenfack, E.; Yang, W.-Y.; Kerr, S.M.; Koeck, T.; Campbell, A.; Kuznetsova, T.; Mischak, H.; Padmanabhan, S.; et al. Novel urinary peptidomic classifier predicts incident heart failure. J. Am. Heart Assoc. 2017, 6, e005432.
- Di Meo, A.; Bartruch, I.; Yousef, A.G.; Pasic, M.D.; Diamandis, E.P.; Yousef, G.M. An integrated proteomic and peptidomic assessment of the normal human urinome. Clin. Chem. Lab. Med. 2017, 55, 237.
- Osorio, D.; Rondon-Villarreal, P.; Torres, R. Peptides: A package for data mining of antimicrobial peptides. R J. 2015, 7, 4–14.
- Klein, J.; Eales, J.; Zürbig, P.; Vlahou, A.; Mischak, H.; Stevens, R. Proteasix: A tool for automated and large-scale prediction of proteases involved in naturally occurring peptide generation. Proteomics 2013, 13, 1077–1082.
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632.
- Martin-Lorenzo, M.; Zubiri, I.; Maroto, A.S.; Gonzalez-Calero, L.; Posada-Ayala, M.; de la Cuesta, F.; Mourino-Alvarez, L.; Lopez-Almodovar, L.F.; Calvo-Bonacho, E.; Ruilope, L.M.; et al. KLK1 and ZG16B proteins and arginine-proline metabolism identified as novel targets to monitor atherosclerosis, acute coronary syndrome and recovery. Metabolomics 2015, 11, 1056–1067.
- Oe, Y.; Hayashi, S.; Fushima, T.; Sato, E.; Kisu, K.; Sato, H.; Ito, S.; Takahashi, N. Coagulation factor Xa and protease-activated receptor 2 as novel therapeutic targets for diabetic nephropathy. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1525–1533.
- Bohl, D.L.; Brennan, D.C. BK virus nephropathy and kidney transplantation. Clin. J. Am. Soc. Nephrol. 2007, 2, S36–S46.
- Tao, Y.; Kim, J.; Stanley, M.; He, Z.; Faubel, S.; Schrier, R.W.; Edelstein, C.L. Pathways of caspase-mediated apoptosis in autosomal-dominant polycystic kidney disease (ADPKD). Kidney Int. 2005, 67, 909–919.
- Gerner, C.; Costigliola, V.; Golubnitschaja, O. Multiomic patterns in body fluids: Technological challenge with a great potential to implement the advanced paradigm of 3P medicine. Mass Spectrom. Rev. 2020, 39, 442–451.