DNA repeat expansion disorders are a group of neuromuscular and neurodegenerative diseases that arise from the inheritance of long tracts of nucleotide repetitions, located in the regulatory region, introns, or inside the coding sequence of a gene. Although loss of protein expression and/or the gain of function of its transcribed mRNA or translated product represent the major pathogenic effect of these pathologies, mitochondrial dysfunction and imbalance in redox homeostasis are reported as common features in these disorders, deeply affecting their severity and progression.
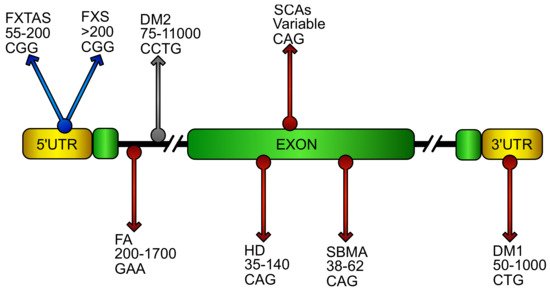
- DNA repeat expansion disorders
- NRF2
- oxidative stress
- FXTAS
- fragile X syndrome
- Friedreich’s ataxia
- myotonic dystrophy
- spinocerebellar ataxia
- Huntington’s disease
- spinal and bulbar muscular atrophy
1. Introduction
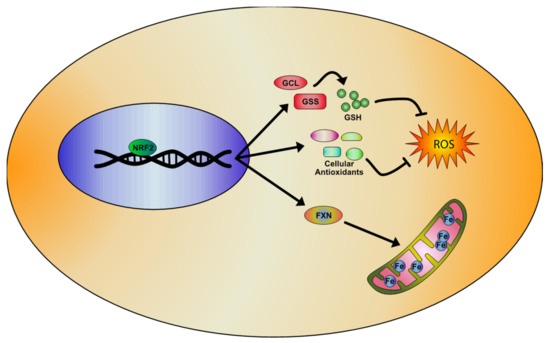
2. NRF2 Pathway and Its Regulation
3. Examples of Oxidative Stress in Loss of Function DNA Expansion Disease
3.1. Friedreich’s Ataxia (FA)
4. Examples of Oxidative Stress in CAG/polyQ diseases
4.1. Huntington’s Disease (HD)
Huntington’s disease (HD) is a progressive, autosomal dominant neurodegenerative disease with defects in the striatum, cerebral cortex, and thalamus [124][125]. The HD disorder is caused by the abnormal expansion of the nucleotide triplet CAG in the gene coding for the protein huntingtin [126]. In the huntingtin gene (HTT) of healthy subjects, the number of trinucleotides CAG repeats varies from 1 to 34, while in HD patients, the CAG triplet expansion ranges between 35–140 repetitions [127]. Clinical features of HD include progressive motor dysfunction, psychiatric disturbance, cognitive decline, dystonia, bradykinesia, and dementia, ultimately leading to death within approximately 15–20 years from the age of onset [128]. The genetic abnormality in the HD gene leads to the formation of a mutant huntingtin protein (mHtt), which is normally involved in the vesicle transport and represents a scaffold for the autophagic machinery [129][130]. The mutant protein exhibits toxic properties, leading to protein aggregation, transcriptional dysregulation, defective energy metabolism, chronic inflammation, and oxidative stress [131][132][133][134]. Inflammation, mitochondrial dysfunction, and oxidative stress are some of the key pathways persistently abnormal in mouse models of HD and in autoptic tissues of patients.
Several pharmacological HD mice models have been developed, resembling defective neuro-motor functions described in human HD patients [135][136][137][138] and supporting oxidative damage as a pathogenic mechanism underlying neurodegeneration in this disease [139][140]. Increased markers of oxidative stress, mitochondrial failure, and chronic inflammation have been found in brain tissue of HD patients. High levels of malondialdehyde, 8-hydroxy-deoxyguanosina, and carbonyls, and lower levels of GSH, SOD1, and GPX, have been detected in plasma and red blood cells of patients [141][142]. Additionally, mitochondrial DNA damage, low levels of oxidative phosphorylation enzymes, and iron-mediated mitochondrial impairment have been shown in autoptic brain tissues of patients [139][143]. In addition, increased amounts of circulating pro-inflammatory cytokines have been reported in patients, whose levels correlated to the severity of the disease [142]. Numerous studies have been focused to reduce oxidative damage in HD by using antioxidants (alpha-tocopherol, CoQ10, vitamin E, vitamin C, N-acetylcysteine (NAC), lipoic acid [144][145][146][147][148][149][150]). Nevertheless, these compounds have shown a moderate effectiveness in counteracting oxidative stress in mouse models, thus leading to hypothesize that a pharmacological upstream activation of NRF2 should be required. Recently, the potent NRF2 inducer SFN has been tested, showing increased mHtt degradation and a significant reduction of cytotoxicity by the NRF2-mediated activation of the ubiquitin–proteasome system [151]. The SFN pre-treatement ameliorated behavioral impairments and reduced pro-inflammatory cytokines in the striatum of a 3-nitropropionic acid (3-NP) mouse model by attenuating neuroinflammation and oxidative stress [151]. High susceptibility to oxidative stress has been also found in human HD neural stem cells, where the genetic correction of the disease-causing mutation restored the redox balance [152]. The protective effect of NRF2 activation in HD patients has been further confirmed in primary monocytes, where the NRF2 induction inhibited the release of pro-inflammatory cytokines (IL-6, IL-1, IL-8, and TNFα) [152]. As in other neurodegenerative diseases, also in HD has been hypothesized a role for ferroptosis in the pathogenic mechanism, mainly as a consequence of increased iron levels that were detected in brain regions of patients [143][153]. Therefore, even in HD, NRF2 can represent a strategic therapeutic target, for its ability in preventing iron overload and regulating ferroptosis-related genes expression [154]. DMF, for instance, exerted beneficial effects on survival and motor functions in R6/2 and YAC128 models of HD, preserving the neuronal integrity in striatum and motor cortex, and slowing degeneration [155]. Additionally, gintonin (GT), a ginseng-derived lysophosphatidic acid receptor ligand, was effective on the NRF2 pathway in the striatum of 3-NPA mice, by protecting the mitochondrial function and reducing the expression of inflammatory mediators (cytokines, COX-2, and iNOS) [156].
5. Examples of Oxidative Stress in RNA Gain of Function Expansion Disease
5.1 Myotonic Dystrophy (DM)
DM is an autosomal dominant disorder, which arises from 2 different mutations: DM1, determined by 50-1000 CUG triplets in the 3′UTR of DMPK gene and DM2, caused by 75– 11000 expansions of the tetranucleotide CCTG in the first intron of ZNF9. DM1 and DM2 are multisystemic diseases sharing a common symptomatology characterized by myotonia, muscular dystrophy, cardiac defects, cataracts [157], and neurological manifestations [158][159]. Unlike DM1, DM2 does not show congenital forms [160]. DM shows a marked somatic instability of repeat expansions that, in DM1, are reported to increase of about 50–80 repeats per year and, in DM2, appear to be even more pronounced [157][161][162]. Depending on the repeat length, the severity of DM1 and the onset of the pathology range from “mild” manifestation (baldness and cataracts) to a “classic” or “juvenile” form, with worse symptoms [19]. On the contrary, although the same clinical heterogeneity is observed in DM2, the pathologic onset and disease severity do not seem to depend on the size of expansions in this disorder [163]. Clinical anticipation, prominent in DM1 [164], appears mildly in DM2 [165].
Different hypotheses have been proposed to explain the pathogenic mechanism in DM. Early studies suggested that the pathological defects observed in DM1 could be determined by the decrease of DMPK expression, mediated by CUG expansions [166] and/or by the trans-acting effect of the expanded mRNA, able to reduce the processing of WT DMPK mRNA [167]. Clinical similarities led to support a common pathogenic mechanism for DM1 and DM2 and, to date, an RNA toxic gain of function is the most credited. In particular, both CUG triplet containing DMPK mRNA and spliced ZNF9 intron1 containing long CCTG sequences are able to sequestrate in the nucleus the splicing factors MBNL1 and 2 [168][169] and, at the same time, to raise the RNA binding activity of CUG-binding protein 1 (CUG-BP1 or CELF1) [170]. This changes the cellular alternative splicing output, determining the defects observed in the disease [171].
Many of the clinical features showed in DM, including myotonia, progressive muscle weakness, cataracts, frontal alopecia, and cognitive decline, suggest an increased susceptibility to oxidative stress in this pathology, as observed in premature and accelerated aging [172]. While in DM2, oxidative stress is still poor investigated, a pathogenic involvement of ROS has been evidenced in DM1. Increased sensitivity to oxidative stress and strong activation of the pro-apoptotic p38 and JNKs pathways have been reported in the C2C12 cell line transfected with human mutant MDPK containing a variable number of CTG repeats [173]. On the contrary, in cells having only 5 CTG repeats, ERKs were preferentially activated [174]. Moreover, studies performed in DM patients have demonstrated an increase of lipid peroxidation and ROS levels, with a parallel decrease of the antioxidant CoQ10 content [175]. Increased oxidative stress and ROS-induced inflammation are known to produce cognitive dysfunctions [176][177] and depressive behaviours [178][179], conditions observed in the MBNL2 KO mouse model of DM1 [180]. In these mice, the chronic administration of methylphenidate (MPH) was able to partially rescue the cognitive defects and depressive-like behaviours, and to reduce the reactive microglia and pro-inflammatory cytokine IL-1β levels [180]. The treatment with MPH was shown to increase NRF2 gene expression in the hippocampus of MBNL2 KO mice and the brain-derived neurotrophic factor (BDNF) levels, which regulates NRF2 nuclear translocation by means of an ERK/PI3K-dependent activation [181]. These findings suggest that the rescue of behavioural defects in MBNL2 KO mice may depend on NRF2-mediated reduction of oxidative stress and inflammation. In line with this, it is important to note that in NRF2 KO mice, an increase of the serum level of pro-inflammatory cytokines and a decrease of the BDNF expression have been reported in association to a depressive-like phenotype [182]. In the same way, NRF2 activation is able to reduce depression and serum content of pro-inflammatory markers induced by lipopolysaccharide (LPS) injections in mice [183]. Cognitive defects [158][159] and depression [184], together with serum increased concentration of the pro-inflammatory IL-6 [185] have been found in DM1 patients, thus the pharmacological NRF2 induction could be very promising in this disease.
References
- Gur-Arie, R.; Cohen, C.J.; Eitan, Y.; Shelef, L.; Hallerman, E.M.; Kashi, Y. Simple sequence repeats in Escherichia coli: Abundance, distribution, composition, and polymorphism. Genome Res. 2000, 10, 62–71.
- Hamada, H.; Petrino, M.G.; Kakunaga, T. A novel repeated element with Z-DNA-forming potential is widely found in evolutionarily diverse eukaryotic genomes. Proc. Natl. Acad. Sci. USA 1982, 79, 6465–6469.
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921.
- Beckman, J.S.; Weber, J.L. Survey of human and rat microsatellites. Genomics 1992, 12, 627–631.
- Toth, G.; Gaspari, Z.; Jurka, J. Microsatellites in different eukaryotic genomes: Survey and analysis. Genome Res. 2000, 10, 967–981.
- Metzgar, D.; Bytof, J.; Wills, C. Selection against frameshift mutations limits microsatellite expansion in coding DNA. Genome Res. 2000, 10, 72–80.
- Bagshaw, A.T.M. Functional Mechanisms of Microsatellite DNA in Eukaryotic Genomes. Genome Biol. Evol. 2017, 9, 2428–2443.
- Hui, J.; Hung, L.H.; Heiner, M.; Schreiner, S.; Neumuller, N.; Reither, G.; Haas, S.A.; Bindereif, A. Intronic CA-repeat and CA-rich elements: A new class of regulators of mammalian alternative splicing. Embo J. 2005, 24, 1988–1998.
- Tseng, S.H.; Cheng, C.Y.; Huang, M.Z.; Chung, M.Y.; Su, T.S. Modulation of formation of the 3′-end of the human argininosuccinate synthetase mRNA by GT-repeat polymorphism. Int. J. Biochem. Mol. Biol. 2013, 4, 179–190.
- Kramer, M.; Sponholz, C.; Slaba, M.; Wissuwa, B.; Claus, R.A.; Menzel, U.; Huse, K.; Platzer, M.; Bauer, M. Alternative 5′ untranslated regions are involved in expression regulation of human heme oxygenase-1. PLoS ONE 2013, 8, e77224.
- Liu, H.; Mulholland, N.; Fu, H.; Zhao, K. Cooperative activity of BRG1 and Z-DNA formation in chromatin remodeling. Mol. Cell. Biol. 2006, 26, 2550–2559.
- Quilez, J.; Guilmatre, A.; Garg, P.; Highnam, G.; Gymrek, M.; Erlich, Y.; Joshi, R.S.; Mittelman, D.; Sharp, A.J. Polymorphic tandem repeats within gene promoters act as modifiers of gene expression and DNA methylation in humans. Nucleic Acids Res. 2016, 44, 3750–3762.
- Ellegren, H. Microsatellite mutations in the germline: Implications for evolutionary inference. Trends Genet. 2000, 16, 551–558.
- Chistiakov, D.A.; Hellemans, B.; Volckaert, F.A.M. Microsatellites and their genomic distribution, evolution, function and applications: A review with special reference to fish genetics. Aquaculture 2006, 255, 1–29.
- Weissenbach, J. Microsatellite polymorphisms and the genetic linkage map of the human genome. Curr. Opin. Genet. Dev. 1993, 3, 414–417.
- López Castel, A.; Cleary, J.D.; Pearson, C.E. Repeat instability as the basis for human diseases and as a potential target for therapy. Nat. Rev. Mol. Cell Biol. 2010, 11, 165–170.
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742.
- Gatchel, J.R.; Zoghbi, H.Y. Diseases of unstable repeat expansion: Mechanisms and common principles. Nat. Rev. Genet. 2005, 6, 743–755.
- Paulson, H. Repeat expansion diseases. Handb. Clin. Neurol. 2018, 147, 105–123.
- McInnis, M.G. Anticipation: An old idea in new genes. Am. J. Hum. Genet. 1996, 59, 973–979.
- Van Mossevelde, S.; van der Zee, J.; Gijselinck, I.; Sleegers, K.; De Bleecker, J.; Sieben, A.; Vandenberghe, R.; Van Langenhove, T.; Baets, J.; Deryck, O.; et al. Clinical Evidence of Disease Anticipation in Families Segregating a C9orf72 Repeat Expansion. JAMA Neurol. 2017, 74, 445–452.
- Paulson, H.L.; Fischbeck, K.H. Trinucleotide repeats in neurogenetic disorders. Annu. Rev. Neurosci. 1996, 19, 79–107.
- Reetz, K.; Dogan, I.; Costa, A.S.; Dafotakis, M.; Fedosov, K.; Giunti, P.; Parkinson, M.H.; Sweeney, M.G.; Mariotti, C.; Panzeri, M.; et al. Biological and clinical characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS) cohort: A cross-sectional analysis of baseline data. Lancet Neurol. 2015, 14, 174–182.
- Cook, A.; Giunti, P. Friedreich’s ataxia: Clinical features, pathogenesis and management. Br. Med Bull. 2017, 124, 19–30.
- Andrew, S.E.; Goldberg, Y.P.; Kremer, B.; Telenius, H.; Theilmann, J.; Adam, S.; Starr, E.; Squitieri, F.; Lin, B.; Kalchman, M.A.; et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington’s disease. Nat. Genet. 1993, 4, 398–403.
- Figueroa, K.P.; Coon, H.; Santos, N.; Velazquez, L.; Mederos, L.A.; Pulst, S.-M. Genetic analysis of age at onset variation in spinocerebellar ataxia type 2. Neurol. Genet. 2017, 3, e155.
- Igarashi, S.; Tanno, Y.; Onodera, O.; Yamazaki, M.; Sato, S.; Ishikawa, A.; Miyatani, N.; Nagashima, M.; Ishikawa, Y.; Sahashi, K.; et al. Strong correlation between the number of CAG repeats in androgen receptor genes and the clinical onset of features of spinal and bulbar muscular atrophy. Neurology 1992, 42, 2300–2302.
- Savić, D.; Rakocvic-Stojanovic, V.; Keckarevic, D.; Culjkovic, B.; Stojkovic, O.; Mladenovic, J.; Todorovic, S.; Apostolski, S.; Romac, S. 250 CTG repeats in DMPK is a threshold for correlation of expansion size and age at onset of juvenile-adult DM1. Hum. Mutat. 2002, 19, 131–139.
- Leehey, M.A.; Berry-Kravis, E.; Goetz, C.G.; Zhang, L.; Hall, D.A.; Li, L.; Rice, C.D.; Lara, R.; Cogswell, J.; Reynolds, A.; et al. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology 2008, 70, 1397–1402.
- Delatycki, M.B.; Paris, D.B.; Gardner, R.J.; Nicholson, G.A.; Nassif, N.; Storey, E.; MacMillan, J.C.; Collins, V.; Williamson, R.; Forrest, S.M. Clinical and genetic study of Friedreich ataxia in an Australian population. Am. J. Med. Genet. 1999, 87, 168–174.
- Cossée, M.; Dürr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschütter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206.
- De Michele, G.; Perrone, F.; Filla, A.; Mirante, E.; Giordano, M.; De Placido, S.; Campanella, G. Age of onset, sex, and cardiomyopathy as predictors of disability and survival in Friedreich’s disease: A retrospective study on 119 patients. Neurology 1996, 47, 1260–1264.
- Cnop, M.; Mulder, H.; Igoillo-Esteve, M. Diabetes in Friedreich ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 94–102.
- Arsenault, M.E.; Prévost, C.; Lescault, A.; Laberge, C.; Puymirat, J.; Mathieu, J. Clinical characteristics of myotonic dystrophy type 1 patients with small CTG expansions. Neurology 2006, 66, 1248–1250.
- Feng, Y.; Zhang, F.; Lokey, L.K.; Chastain, J.L.; Lakkis, L.; Eberhart, D.; Warren, S.T. Translational suppression by trinucleotide repeat expansion at FMR1. Science 1995, 268, 731–734.
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427.
- Zoghbi, H.Y.; Orr, H.T. Glutamine repeats and neurodegeneration. Annu. Rev. Neurosci. 2000, 23, 217–247.
- Nucifora, F.C., Jr.; Sasaki, M.; Peters, M.F.; Huang, H.; Cooper, J.K.; Yamada, M.; Takahashi, H.; Tsuji, S.; Troncoso, J.; Dawson, V.L.; et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science 2001, 291, 2423–2428.
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.-M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 2002, 296, 2238–2243.
- Ciechanover, A.; Brundin, P. The ubiquitin proteasome system in neurodegenerative diseases: Sometimes the chicken, sometimes the egg. Neuron 2003, 40, 427–446.
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barceló, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255.
- Ranum, L.P.; Rasmussen, P.F.; Benzow, K.A.; Koob, M.D.; Day, J.W. Genetic mapping of a second myotonic dystrophy locus. Nat. Genet. 1998, 19, 196–198.
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.; Grigsby, J.; Zhang, L.; Brunberg, J.A.; Greco, C.; Des Portes, V.; Jardini, T.; Levine, R.; et al. Fragile X premutation tremor/ataxia syndrome: Molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet. 2003, 72, 869–878.
- Echeverria, G.V.; Cooper, T.A. Muscleblind-like 1 activates insulin receptor exon 11 inclusion by enhancing U2AF65 binding and splicing of the upstream intron. Nucleic Acids Res. 2014, 42, 1893–1903.
- Savkur, R.S.; Philips, A.V.; Cooper, T.A. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat. Genet. 2001, 29, 40–47.
- Mankodi, A.; Takahashi, M.P.; Jiang, H.; Beck, C.L.; Bowers, W.J.; Moxley, R.T.; Cannon, S.C.; Thornton, C.A. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol. Cell 2002, 10, 35–44.
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010, 29, 1248–1261.
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The Role of Oxidative Stress in Neurodegenerative Diseases. Exp. Neurobiol. 2015, 24, 325–340.
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967.
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930.
- Amoutzias, G.D.; Robertson, D.L.; Van de Peer, Y.; Oliver, S.G. Choose your partners: Dimerization in eukaryotic transcription factors. Trends Biochem. Sci. 2008, 33, 220–229.
- Igarashi, K.; Kataoka, K.; Itoh, K.; Hayashi, N.; Nishizawa, M.; Yamamoto, M. Regulation of transcription by dimerization of erythroid factor NF-E2 p45 with small Maf proteins. Nature 1994, 367, 568–572.
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322.
- Wasserman, W.W.; Fahl, W.E. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. USA 1997, 94, 5361–5366.
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317.
- Kasai, S.; Shimizu, S.; Tatara, Y.; Mimura, J.; Itoh, K. Regulation of Nrf2 by Mitochondrial Reactive Oxygen Species in Physiology and Pathology. Biomolecules 2020, 10, 320.
- Corenblum, M.J.; Ray, S.; Remley, Q.W.; Long, M.; Harder, B.; Zhang, D.D.; Barnes, C.A.; Madhavan, L. Reduced Nrf2 expression mediates the decline in neural stem cell function during a critical middle-age period. Aging Cell 2016, 15, 725–736.
- Dodson, M.; de la Vega, M.R.; Cholanians, A.B.; Schmidlin, C.J.; Chapman, E.; Zhang, D.D. Modulating NRF2 in Disease: Timing Is Everything. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 555–575.
- La Rosa, P.; Russo, M.; D’Amico, J.; Petrillo, S.; Aquilano, K.; Lettieri-Barbato, D.; Turchi, R.; Bertini, E.S.; Piemonte, F. Nrf2 Induction Re-establishes a Proper Neuronal Differentiation Program in Friedreich’s Ataxia Neural Stem Cells. Front. Cell. Neurosci. 2019, 13, 356.
- Robledinos-Anton, N.; Rojo, A.I.; Ferreiro, E.; Nunez, A.; Krause, K.H.; Jaquet, V.; Cuadrado, A. Transcription factor NRF2 controls the fate of neural stem cells in the subgranular zone of the hippocampus. Redox Biol. 2017, 13, 393–401.
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51.
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 pathway and its cross talk with Nrf2 in mitochondrial quality control. J. Clin. Biochem. Nutr. 2019, 64, 1–12.
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 Signaling Network Defines Clinical Biomarkers and Therapeutic Opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916.
- Petrillo, S.; D’Amico, J.; La Rosa, P.; Bertini, E.S.; Piemonte, F. Targeting NRF2 for the Treatment of Friedreich’s Ataxia: A Comparison among Drugs. Int. J. Mol. Sci. 2019, 20, 5211.
- Cuadrado, A. Structural and functional characterization of Nrf2 degradation by glycogen synthase kinase 3/beta-TrCP. Free Radic. Biol. Med. 2015, 88, 147–157.
- Kobayashi, A.; Kang, M.I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139.
- Shaw, P.; Chattopadhyay, A. Nrf2-ARE signaling in cellular protection: Mechanism of action and the regulatory mechanisms. J. Cell. Physiol. 2019, 235, 3119–3130.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203.
- Rada, P.; Rojo, A.I.; Chowdhry, S.; McMahon, M.; Hayes, J.D.; Cuadrado, A. SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell. Biol. 2011, 31, 1121–1133.
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267.
- Liu, Y.; Pang, Y.; Caisova, V.; Ding, J.; Yu, D.; Zhou, Y.; Huynh, T.T.; Ghayee, H.; Pacak, K.; Yang, C. Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma. Cancers 2020, 12, 280.
- Liu, Y.; Lu, Y.; Celiku, O.; Li, A.; Wu, Q.; Zhou, Y.; Yang, C. Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J. Natl. Cancer Inst. 2019, 111, 1033–1041.
- Benarroch, E.E. Nrf2, cellular redox regulation, and neurologic implications. Neurology 2017, 88, 1942–1950.
- Dinkova-Kostova, A.T.; Kostov, R.V.; Kazantsev, A.G. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J. 2018, 285, 3576–3590.
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466.
- Basak, P.; Sadhukhan, P.; Sarkar, P.; Sil, P.C. Perspectives of the Nrf-2 signaling pathway in cancer progression and therapy. Toxicol. Rep. 2017, 4, 306–318.
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of NRF2 in Cancer. Trends Mol. Med. 2016, 22, 578–593.
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85.
- Kitamura, H.; Motohashi, H. NRF2 addiction in cancer cells. Cancer Sci. 2018, 109, 900–911.
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85.
- Sarlette, A.; Krampfl, K.; Grothe, C.; Neuhoff, N.; Dengler, R.; Petri, S. Nuclear erythroid 2-related factor 2-antioxidative response element signaling pathway in motor cortex and spinal cord in amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2008, 67, 1055–1062.
- Brandes, M.S.; Gray, N.E. NRF2 as a Therapeutic Target in Neurodegenerative Diseases. ASN Neuro 2020, 12, 1759091419899782.
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Redox Mechanisms in Neurodegeneration: From Disease Outcomes to Therapeutic Opportunities. Antioxid. Redox Signal. 2019, 30, 1450–1499.
- Cossée, M.; Schmitt, M.; Campuzano, V.; Reutenauer, L.; Moutou, C.; Mandel, J.L.; Koenig, M. Evolution of the Friedreich’s ataxia trinucleotide repeat expansion: Founder effect and premutations. Proc. Natl. Acad. Sci. USA 1997, 94, 7452–7457.
- Kim, E.; Napierala, M.; Dent, S.Y. Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich’s ataxia. Nucleic Acids Res. 2011, 39, 8366–8377.
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12.
- Pandolfo, M.; Pastore, A. The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J. Neurol. 2009, 256 (Suppl. 1), 9–17.
- Parkinson, M.H.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Giunti, P. Clinical features of Friedreich’s ataxia: Classical and atypical phenotypes. J. Neurochem. 2013, 126 (Suppl. 1), 103–117.
- Puccio, H.; Simon, D.; Cossee, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186.
- Anzovino, A.; Lane, D.J.; Huang, M.L.; Richardson, D.R. Fixing frataxin: ‘ironing out’ the metabolic defect in Friedreich’s ataxia. Br. J. Pharmacol. 2014, 171, 2174–2190.
- Vaubel, R.A.; Isaya, G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 2013, 55, 50–61.
- Koenig, M.; Mandel, J.L. Deciphering the cause of Friedreich ataxia. Curr. Opin. Neurobiol. 1997, 7, 689–694.
- Gomes, C.M.; Santos, R. Neurodegeneration in Friedreich’s ataxia: From defective frataxin to oxidative stress. Oxid. Med. Cell. Longev. 2013, 2013, 487534.
- Carletti, B.; Piemonte, F. Friedreich’s Ataxia: A Neuronal Point of View on the Oxidative Stress Hypothesis. Antioxidants 2014, 3, 592–603.
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727.
- Anzovino, A.; Chiang, S.; Brown, B.E.; Hawkins, C.L.; Richardson, D.R.; Huang, M.L. Molecular Alterations in a Mouse Cardiac Model of Friedreich Ataxia: An Impaired Nrf2 Response Mediated via Upregulation of Keap1 and Activation of the Gsk3β Axis. Am. J. Pathol. 2017, 187, 2858–2875.
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchere, F.; Lonn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253.
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865.
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493.
- Emond, M.; Lepage, G.; Vanasse, M.; Pandolfo, M. Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 2000, 55, 1752–1753.
- Schulz, J.B.; Dehmer, T.; Schöls, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurology 2000, 55, 1719–1721.
- Tozzi, G.; Nuccetelli, M.; Lo Bello, M.; Bernardini, S.; Bellincampi, L.; Ballerini, S.; Gaeta, L.M.; Casali, C.; Pastore, A.; Federici, G.; et al. Antioxidant enzymes in blood of patients with Friedreich’s ataxia. Arch. Dis. Child. 2002, 86, 376–379.
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440.
- Cotticelli, M.G.; Crabbe, A.M.; Wilson, R.B.; Shchepinov, M.S. Insights into the role of oxidative stress in the pathology of Friedreich ataxia using peroxidation resistant polyunsaturated fatty acids. Redox Biol. 2013, 1, 398–404.
- Cotticelli, M.G.; Xia, S.; Lin, D.; Lee, T.; Terrab, L.; Wipf, P.; Huryn, D.M.; Wilson, R.B. Ferroptosis as a Novel Therapeutic Target for Friedreich’s Ataxia. J. Pharmacol. Exp. Ther. 2019, 369, 47–54.
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. ‘Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia’. Cell Death Dis. 2016, 7, e2237.
- Kajarabille, N.; Latunde-Dada, G.O. Programmed Cell-Death by Ferroptosis: Antioxidants as Mitigators. Int. J. Mol. Sci. 2019, 20.
- Esteras, N.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 activation in the treatment of neurodegenerative diseases: A focus on its role in mitochondrial bioenergetics and function. Biol. Chem. 2016, 397, 383–400.
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116.
- Klomparens, E.A.; Ding, Y. The neuroprotective mechanisms and effects of sulforaphane. Brain Circ. 2019, 5, 74–83.
- Uddin, M.S.; Mamun, A.A.; Jakaria, M.; Thangapandiyan, S.; Ahmad, J.; Rahman, M.A.; Mathew, B.; Abdel-Daim, M.M.; Aleya, L. Emerging promise of sulforaphane-mediated Nrf2 signaling cascade against neurological disorders. Sci. Total Environ. 2020, 707, 135624.
- Scannevin, R.H.; Chollate, S.; Jung, M.Y.; Shackett, M.; Patel, H.; Bista, P.; Zeng, W.; Ryan, S.; Yamamoto, M.; Lukashev, M.; et al. Fumarates promote cytoprotection of central nervous system cells against oxidative stress via the nuclear factor (erythroid-derived 2)-like 2 pathway. J. Pharmacol. Exp. Ther. 2012, 341, 274–284.
- Montes Diaz, G.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250.
- Ranea-Robles, P.; Launay, N.; Ruiz, M.; Calingasan, N.Y.; Dumont, M.; Naudi, A.; Portero-Otin, M.; Pamplona, R.; Ferrer, I.; Beal, M.F.; et al. Aberrant regulation of the GSK-3beta/NRF2 axis unveils a novel therapy for adrenoleukodystrophy. EMBO Mol. Med. 2018, 10, e8604.
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173.
- Jasoliya, M.; Sacca, F.; Sahdeo, S.; Chedin, F.; Pane, C.; Brescia Morra, V.; Filla, A.; Pook, M.; Cortopassi, G. Dimethyl fumarate dosing in humans increases frataxin expression: A potential therapy for Friedreich’s Ataxia. PLoS ONE 2019, 14, e0217776.
- Sahdeo, S.; Scott, B.D.; McMackin, M.Z.; Jasoliya, M.; Brown, B.; Wulff, H.; Perlman, S.L.; Pook, M.A.; Cortopassi, G.A. Dyclonine rescues frataxin deficiency in animal models and buccal cells of patients with Friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 6848–6862.
- Clay, A.; Hearle, P.; Schadt, K.; Lynch, D.R. New developments in pharmacotherapy for Friedreich ataxia. Expert Opin. Pharmacother. 2019, 20, 1855–1867.
- Hausse, A.O.; Aggoun, Y.; Bonnet, D.; Sidi, D.; Munnich, A.; Rotig, A.; Rustin, P. Idebenone and reduced cardiac hypertrophy in Friedreich’s ataxia. Heart 2002, 87, 346–349.
- Di Prospero, N.A.; Baker, A.; Jeffries, N.; Fischbeck, K.H. Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: A randomised, placebo-controlled trial. Lancet Neurol. 2007, 6, 878–886.
- Montenegro, L.; Turnaturi, R.; Parenti, C.; Pasquinucci, L. Idebenone: Novel Strategies to Improve Its Systemic and Local Efficacy. Nanomaterials 2018, 8, 87.
- Lynch, D.R.; Farmer, J.; Hauser, L.; Blair, I.A.; Wang, Q.Q.; Mesaros, C.; Snyder, N.; Boesch, S.; Chin, M.; Delatycki, M.B.; et al. Safety, pharmacodynamics, and potential benefit of omaveloxolone in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2019, 6, 15–26.
- Zesiewicz, T.; Salemi, J.L.; Perlman, S.; Sullivan, K.L.; Shaw, J.D.; Huang, Y.; Isaacs, C.; Gooch, C.; Lynch, D.R.; Klein, M.B. Double-blind, randomized and controlled trial of EPI-743 in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2018, 8, 233–242.
- Furtado, S.; Suchowersky, O.; Rewcastle, B.; Graham, L.; Klimek, M.L.; Garber, A. Relationship between trinucleotide repeats and neuropathological changes in Huntington’s disease. Ann. Neurol. 1996, 39, 132–136.
- Snell, R.G.; MacMillan, J.C.; Cheadle, J.P.; Fenton, I.; Lazarou, L.P.; Davies, P.; MacDonald, M.E.; Gusella, J.F.; Harper, P.S.; Shaw, D.J. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Genet. 1993, 4, 393–397.
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983.
- Ravina, B.; Romer, M.; Constantinescu, R.; Biglan, K.; Brocht, A.; Kieburtz, K.; Shoulson, I.; McDermott, M.P. The relationship between CAG repeat length and clinical progression in Huntington’s disease. Mov. Disord. 2008, 23, 1223–1227.
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228.
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894.
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275.
- Shacham, T.; Sharma, N.; Lederkremer, G.Z. Protein Misfolding and ER Stress in Huntington’s Disease. Front. Mol. Biosci. 2019, 6, 20.
- Gallardo-Orihuela, A.; Hervás-Corpión, I.; Hierro-Bujalance, C.; Sanchez-Sotano, D.; Jiménez-Gómez, G.; Mora-López, F.; Campos-Caro, A.; Garcia-Alloza, M.; Valor, L.M. Transcriptional correlates of the pathological phenotype in a Huntington’s disease mouse model. Sci. Rep. 2019, 9, 18696.
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 2014, 17, 822–831.
- Stack, E.C.; Matson, W.R.; Ferrante, R.J. Evidence of oxidant damage in Huntington’s disease: Translational strategies using antioxidants. Ann. N. Y. Acad. Sci. 2008, 1147, 79–92.
- Agrawal, S.; Fox, J.H. Novel proteomic changes in brain mitochondria provide insights into mitochondrial dysfunction in mouse models of Huntington’s disease. Mitochondrion 2019, 47, 318–329.
- Bogdanov, M.B.; Ferrante, R.J.; Kuemmerle, S.; Klivenyi, P.; Beal, M.F. Increased vulnerability to 3-nitropropionic acid in an animal model of Huntington’s disease. J. Neurochem. 1998, 71, 2642–2644.
- Brouillet, E. The 3-NP Model of Striatal Neurodegeneration. Curr. Protoc. Neurosci. 2014, 67, 9.48.1–9.48.14.
- Rosenstock, T.R.; Carvalho, A.C.; Jurkiewicz, A.; Frussa-Filho, R.; Smaili, S.S. Mitochondrial calcium, oxidative stress and apoptosis in a neurodegenerative disease model induced by 3-nitropropionic acid. J. Neurochem. 2004, 88, 1220–1228.
- Browne, S.E.; Beal, M.F. Oxidative damage in Huntington’s disease pathogenesis. Antioxid. Redox Signal. 2006, 8, 2061–2073.
- Maiuri, T.; Bowie, L.E.; Truant, R. DNA Repair Signaling of Huntingtin: The Next Link Between Late-Onset Neurodegenerative Disease and Oxidative DNA Damage. DNA Cell Biol. 2019, 38, 1–6.
- Chen, C.M.; Wu, Y.R.; Cheng, M.L.; Liu, J.L.; Lee, Y.M.; Lee, P.W.; Soong, B.W.; Chiu, D.T. Increased oxidative damage and mitochondrial abnormalities in the peripheral blood of Huntington’s disease patients. Biochem. Biophys. Res. Commun. 2007, 359, 335–340.
- Sánchez-López, F.; Tasset, I.; Agüera, E.; Feijóo, M.; Fernández-Bolaños, R.; Sánchez, F.M.; Ruiz, M.C.; Cruz, A.H.; Gascón, F.; Túnez, I. Oxidative stress and inflammation biomarkers in the blood of patients with Huntington’s disease. Neurol. Res. 2012, 34, 721–724.
- Agrawal, S.; Fox, J.; Thyagarajan, B.; Fox, J.H. Brain mitochondrial iron accumulates in Huntington’s disease, mediates mitochondrial dysfunction, and can be removed pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329.
- Peyser, C.E.; Folstein, M.; Chase, G.A.; Starkstein, S.; Brandt, J.; Cockrell, J.R.; Bylsma, F.; Coyle, J.T.; McHugh, P.R.; Folstein, S.E. Trial of d-alpha-tocopherol in Huntington’s disease. Am. J. Psychiatry 1995, 152, 1771–1775.
- Kasparová, S.; Sumbalová, Z.; Bystrický, P.; Kucharská, J.; Liptaj, T.; Mlynárik, V.; Gvozdjáková, A. Effect of coenzyme Q10 and vitamin E on brain energy metabolism in the animal model of Huntington’s disease. Neurochem. Int. 2006, 48, 93–99.
- Rebec, G.V.; Barton, S.J.; Marseilles, A.M.; Collins, K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport 2003, 14, 1263–1265.
- Wright, D.J.; Gray, L.J.; Finkelstein, D.I.; Crouch, P.J.; Pow, D.; Pang, T.Y.; Li, S.; Smith, Z.M.; Francis, P.S.; Renoir, T.; et al. N-acetylcysteine modulates glutamatergic dysfunction and depressive behavior in Huntington’s disease. Hum. Mol. Genet. 2016, 25, 2923–2933.
- Wright, D.J.; Renoir, T.; Smith, Z.M.; Frazier, A.E.; Francis, P.S.; Thorburn, D.R.; McGee, S.L.; Hannan, A.J.; Gray, L.J. N-Acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington’s disease. Transl. Psychiatry 2015, 5, e492.
- Sandhir, R.; Sood, A.; Mehrotra, A.; Kamboj, S.S. N-Acetylcysteine reverses mitochondrial dysfunctions and behavioral abnormalities in 3-nitropropionic acid-induced Huntington’s disease. Neurodegener. Dis. 2012, 9, 145–157.
- Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Beal, M.F. Lipoic acid improves survival in transgenic mouse models of Huntington’s disease. Neuroreport 2001, 12, 3371–3373.
- Liu, Y.; Hettinger, C.L.; Zhang, D.; Rezvani, K.; Wang, X.; Wang, H. Sulforaphane enhances proteasomal and autophagic activities in mice and is a potential therapeutic reagent for Huntington’s disease. J. Neurochem. 2014, 129, 539–547.
- Quinti, L.; Dayalan Naidu, S.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-modifying small molecule reveals muted NRF2 signaling responses in neural stem cells from Huntington’s disease patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685.
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556.
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107.
- Ellrichmann, G.; Petrasch-Parwez, E.; Lee, D.H.; Reick, C.; Arning, L.; Saft, C.; Gold, R.; Linker, R.A. Efficacy of fumaric acid esters in the R6/2 and YAC128 models of Huntington’s disease. PLoS ONE 2011, 6, e16172.
- Jang, M.; Choi, J.H.; Chang, Y.; Lee, S.J.; Nah, S.Y.; Cho, I.H. Gintonin, a ginseng-derived ingredient, as a novel therapeutic strategy for Huntington’s disease: Activation of the Nrf2 pathway through lysophosphatidic acid receptors. Brain Behav. Immun. 2019, 80, 146–162.
- Schoser, B.; Timchenko, L. Myotonic dystrophies 1 and 2: Complex diseases with complex mechanisms. Curr. Genom. 2010, 11, 77–90.
- Winblad, S.; Samuelsson, L.; Lindberg, C.; Meola, G. Cognition in myotonic dystrophy type 1: A 5-year follow-up study. Eur. J. Neurol. 2016, 23, 1471–1476.
- Sansone, V.; Gandossini, S.; Cotelli, M.; Calabria, M.; Zanetti, O.; Meola, G. Cognitive impairment in adult myotonic dystrophies: A longitudinal study. Neurol. Sci. 2007, 28, 9–15.
- Schara, U.; Schoser, B.G. Myotonic dystrophies type 1 and 2: A summary on current aspects. Semin. Pediatr. Neurol. 2006, 13, 71–79.
- Machuca-Tzili, L.; Brook, D.; Hilton-Jones, D. Clinical and molecular aspects of the myotonic dystrophies: A review. Muscle Nerve 2005, 32, 1–18.
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867.
- Day, J.W.; Ricker, K.; Jacobsen, J.F.; Rasmussen, L.J.; Dick, K.A.; Kress, W.; Schneider, C.; Koch, M.C.; Beilman, G.J.; Harrison, A.R.; et al. Myotonic dystrophy type 2: Molecular, diagnostic and clinical spectrum. Neurology 2003, 60, 657–664.
- Yum, K.; Wang, E.T.; Kalsotra, A. Myotonic dystrophy: Disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr. Opin. Genet. Dev. 2017, 44, 30–37.
- Schneider, C.; Ziegler, A.; Ricker, K.; Grimm, T.; Kress, W.; Reimers, C.D.; Meinck, H.; Reiners, K.; Toyka, K.V. Proximal myotonic myopathy: Evidence for anticipation in families with linkage to chromosome 3q. Neurology 2000, 55, 383–388.
- Reddy, S.; Smith, D.B.; Rich, M.M.; Leferovich, J.M.; Reilly, P.; Davis, B.M.; Tran, K.; Rayburn, H.; Bronson, R.; Cros, D.; et al. Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat. Genet. 1996, 13, 325–335.
- Wang, J.; Pegoraro, E.; Menegazzo, E.; Gennarelli, M.; Hoop, R.C.; Angelini, C.; Hoffman, E.P. Myotonic dystrophy: Evidence for a possible dominant-negative RNA mutation. Hum. Mol. Genet. 1995, 4, 599–606.
- Warf, M.B.; Berglund, J.A. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA 2007, 13, 2238–2251.
- Pagliarini, V.; La Rosa, P.; Sette, C. Faulty RNA splicing: Consequences and therapeutic opportunities in brain and muscle disorders. Hum. Genet. 2017, 136, 1215–1235.
- Kuyumcu-Martinez, N.M.; Wang, G.S.; Cooper, T.A. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell 2007, 28, 68–78.
- Charlet, B.N.; Savkur, R.S.; Singh, G.; Philips, A.V.; Grice, E.A.; Cooper, T.A. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol. Cell 2002, 10, 45–53.
- Mateos-Aierdi, A.J.; Goicoechea, M.; Aiastui, A.; Fernández-Torrón, R.; Garcia-Puga, M.; Matheu, A.; López de Munain, A. Muscle wasting in myotonic dystrophies: A model of premature aging. Front. Aging Neurosci. 2015, 7, 125.
- Usuki, F.; Ishiura, S. Expanded CTG repeats in myotonin protein kinase increase susceptibility to oxidative stress. Neuroreport 1998, 9, 2291–2296.
- Usuki, F.; Takahashi, N.; Sasagawa, N.; Ishiura, S. Differential signaling pathways following oxidative stress in mutant myotonin protein kinase cDNA-transfected C2C12 cell lines. Biochem. Biophys. Res. Commun. 2000, 267, 739–743.
- Ihara, Y.; Mori, A.; Hayabara, T.; Namba, R.; Nobukuni, K.; Sato, K.; Miyata, S.; Edamatsu, R.; Liu, J.; Kawai, M. Free radicals, lipid peroxides and antioxidants in blood of patients with myotonic dystrophy. J. Neurol. 1995, 242, 119–122.
- Head, E. Oxidative damage and cognitive dysfunction: Antioxidant treatments to promote healthy brain aging. Neurochem. Res. 2009, 34, 670–678.
- Baierle, M.; Nascimento, S.N.; Moro, A.M.; Brucker, N.; Freitas, F.; Gauer, B.; Durgante, J.; Bordignon, S.; Zibetti, M.; Trentini, C.M.; et al. Relationship between inflammation and oxidative stress and cognitive decline in the institutionalized elderly. Oxid. Med. Cell. Longev. 2015, 2015, 804198.
- Michel, T.M.; Pülschen, D.; Thome, J. The role of oxidative stress in depressive disorders. Curr. Pharm. Des. 2012, 18, 5890–5899.
- Rawdin, B.J.; Mellon, S.H.; Dhabhar, F.S.; Epel, E.S.; Puterman, E.; Su, Y.; Burke, H.M.; Reus, V.I.; Rosser, R.; Hamilton, S.P.; et al. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav. Immun. 2013, 31, 143–152.
- Ramon-Duaso, C.; Gener, T.; Consegal, M.; Fernández-Avilés, C.; Gallego, J.J.; Castarlenas, L.; Swanson, M.S.; de la Torre, R.; Maldonado, R.; Puig, M.V.; et al. Methylphenidate Attenuates the Cognitive and Mood Alterations Observed in Mbnl2 Knockout Mice and Reduces Microglia Overexpression. Cereb. Cortex 2018, 29, 2978–2997.
- Bruna, B.; Lobos, P.; Herrera-Molina, R.; Hidalgo, C.; Paula-Lima, A.; Adasme, T. The signaling pathways underlying BDNF-induced Nrf2 hippocampal nuclear translocation involve ROS, RyR-Mediated Ca2+ signals, ERK and PI3K. Biochem. Biophys. Res. Commun. 2018, 505, 201–207.
- Yao, W.; Zhang, J.C.; Ishima, T.; Dong, C.; Yang, C.; Ren, Q.; Ma, M.; Han, M.; Wu, J.; Suganuma, H.; et al. Role of Keap1-Nrf2 signaling in depression and dietary intake of glucoraphanin confers stress resilience in mice. Sci. Rep. 2016, 6, 30659.
- Yao, W.; Zhang, J.-C.; Ishima, T.; Ren, Q.; Yang, C.; Dong, C.; Ma, M.; Saito, A.; Honda, T.; Hashimoto, K. Antidepressant effects of TBE-31 and MCE-1, the novel Nrf2 activators, in an inflammation model of depression. Eur. J. Pharmacol. 2016, 793, 21–27.
- Winblad, S.; Jensen, C.; Månsson, J.-E.; Samuelsson, L.; Lindberg, C. Depression in Myotonic Dystrophy type 1: Clinical and neuronal correlates. Behav. Brain Funct. 2010, 6, 25.
- Wegner, M.; Araszkiewicz, A.; Piorunska-Stolzmann, M.; Wierusz-Wysocka, B.; Zozulinska-Ziolkiewicz, D. Association between IL-6 concentration and diabetes-related variables in DM1 patients with and without microvascular complications. Inflammation 2013, 36, 723–728.