Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 5 by Ron Wang and Version 4 by Ron Wang.
Autosomal recessive polycystic kidney disease (ARPKD) is a rare disorder and one of the most severe forms of polycystic kidney disease, leading to end-stage renal disease (ESRD) in childhood. PKHD1 is the gene that is responsible for the vast majority of ARPKD. However, some cases have been related to a new gene that was recently identified (DZIP1L gene), as well as several ciliary genes that can mimic a ARPKD-like phenotypic spectrum. In addition, a number of molecular pathways involved in the ARPKD pathogenesis and progression were elucidated using cellular and animal models. However, the function of the ARPKD proteins and the molecular mechanism of the disease currently remain incompletely understood.
- ARPKD
- cyst
- rare monogenic disease
- nephrology
1. Introduction
Autosomal recessive polycystic kidney disease (ARPKD) is a severe inherited cystic disease characterized by the combination of bilateral renal cystic disease and congenital hepatic fibrosis. ARPKD manifests perinatally, or in childhood, as an important cause of pediatric morbidity and mortality [1]. ARPKD is a rare disease; an incidence of 1 in 8000 births was calculated in an isolated and inbred population from Finland [2]. However, in the Americas (North, Central, and South), the reported incidence is 1 in 26:485 births, and the annualized prevalence is 1.17 per 100,000 [3]. The widespread prevalence of ARPKD is estimated to be 1 in 20:000 births [4].
ARPKD is the recessive form of a group of heterogeneous monogenic disorders named polycystic kidney disease (PKD). The dominant form, autosomal dominant polycystic kidney disease (ADPKD), has a higher epidemiological prevalence and is typically diagnosed in adults [5].
2. Autosomal Recessive Polycystic Kidney Disease Clinical Presentation
ARPKD is phenotypically highly variable; it can present as a disease of perinatal, neonatal, infantile, juvenile, or young adult-onset disease [6], with no known gender or ethnic bias [7]. Typically, the most severe cases of ARPKD present in the late gestational or neonatal stage, with bilateral massively enlarged and echogenic kidneys with poor corticomedullary differentiation, retained reniform contour, and multiple tiny cysts [8][9][10]. In addition, they can present with oligo- or anhydramnios, resulting in the typical “Potter sequence” phenotype with pulmonary hypoplasia, characteristic facial features, and clubfoot contracted limbs [9][11][12]. In addition to the Potter sequence, the presence of other extrarenal manifestations is not common [13]. There are no documented hepatic phenotypes [14], although some associated, such as abdominal dystocia, have been reported [15].
Detection of severe oligohydramnios is associated with worse outcome due to the high risk of associated pulmonary hypoplasia. Up to 50% of ARPKD neonates die of respiratory insufficiency due to pulmonary hypoplasia and thoracic compression. However, after the perinatal period, survival is high, reaching 1-year and 10-year survival rates of 85% and 82%, respectively [7][8][12]. The patients who survive the perinatal period require extensive care by specialists in internal medicine [16]. Note that prenatal diagnosis and termination of pregnancy are factors to consider in the epidemiology of the disease [15][17]. Another problem derived from kidney enlargement and pulmonary hypoplasia, in addition to early uremia and pulmonary immaturity, includes the difficulty of enteral feeding that could complicate nutrition, requiring persistent nasogastric feeding [11][18].
3. Diagnosis
ARPKD is frequently diagnosed in the prenatal period due to its early and severe manifestations. In prenatal diagnosis, an ultrasound from second/third trimester can detect enlarged, echogenic kidneys, and medullary hyperechogenicity, due to the loss of corticomedullary differentiation and diffusively increased hepatic parenchymal echogenicity with fibrous tissue. The presence of oligohydramnios can make it challenging, so ultrasonography and MRI are required. The finding of microcysts (5–7 mm) was reported in 30% of ARPKD cases, but macrocysts (>10 mm) are rare and could indicate another different ciliopathy. These ultrasound findings are common in other pathologies, like Meckel syndrome, and mild forms of the disease may not be detected by prenatal ultrasounds. In these cases, the genetic test offers the possibility of providing an accurate diagnosis [19][20].
Identifying the PKHD1 gene made it possible to perform the genetic diagnosis by direct DNA sequencing (Sanger method). However, the genetic test for the PKHD1 mutation is complicated due to the large genomic size and the allelic heterogeneity of the disease-associated mutations [21]. However, according to the Genetics Work Group, single-gene testing should be avoided in cases of suspected ARPKD due to its broad overlapping phenotypic spectrum. As an alternative, methods such as next generation sequencing have become of interest as techniques that can simultaneously and efficiently analyze multiple candidate genes in a unique test, at relative low cost. In rare cases, mutations in two genes can even be observed in children with severe neonatal clinical phenotype [4][20][22].
The outcome of the genetic testing is essential for clinical management of comorbidities and complications associated with each disease, allowing informed genetic counselling and, in the future, precision medicine on a more specific basis [4].
4. Differential Diagnosis
The ARPKD phenotype is not only caused by mutations in PKHD1. This makes diagnosis and management, including care during the perinatal period, a difficult task. A number of other recessive and dominant genes need to be considered (Figure 1) [4][23][24].
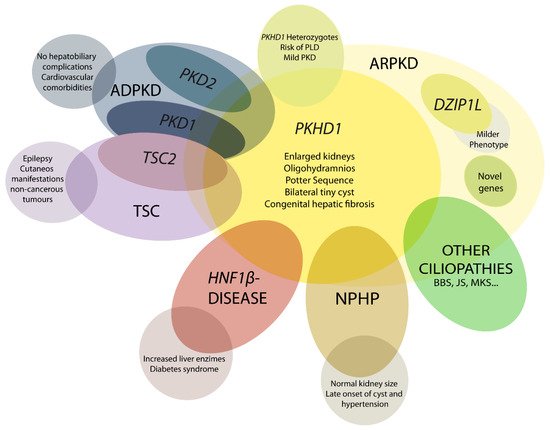
Figure 1. Schematic representation of ARPKD differential diagnosis. PHKD1 is the main causative gene in ARPKD, where DZIP1L present milder phenotype. Mutations in other genes can overlap clinical manifestations of ARPKD, such as PKD1 and PKD2, the main causative genes of autosomal dominant polycystic kidney disease (ADPKD); TSC2, that causes tuberous sclerosis (TSC); and others for instance HNF1β, nephronophthisis (NPHP) genes and other ciliopathies as Bardet–Biedl (BBS), Joubert (JS), and Meckel syndrome (MKS). These overlapping phenotypes manifest the physiologic complex and functional interactions that occur among ciliopathy genes.
5. Genetics of ARPKD
As we mentioned earlier, ARPKD is caused by mutations in PKHD1 and, the recently discovered, DZIP1L [25][26][27]. PKHD1, located on chromosome 6 (6p12.3-p12.2) (Figure 2A) [21][27][28][29], is one of the largest human genes with a genomic segment of ~500 kb. It is predicted to have a minimum of 86 exons assembled in a complicated pattern of alternative splice variants, transcribing a large full-length mRNA of approximately 8.5 kb–13 kb [28]. Multiple types of mutations characterized as pathogenic have been identified across the gene. Currently, approximately 750 PKHD1 mutations have been identified, of which approximately half are missense changes. A missense mutation in exon 3 (c. 107C>T; p.Thr36Me) is the most common mutation described, accounting for more than 20% of all cases [21][30]. This mutation has been observed in the context of heterozygotes, with a second distinct mutant allele [31]. Most cases are familial, but de novo mutations are also reported and account for 2 to 5% of cases [21]. Interestingly, in the context of isolated autosomal dominant polycystic liver disease (ADPLD), Besse and colleagues have reported several individuals with PKHD1 mutations in heterozygote carriers, 10 of 102 ADPLD patients of their cohort were explained by PKHD1 mutations, one of them presented the p.Thr36Me missense variant [32]. According to the clinical observation, it is a genetic fact that 10% of ARPKD patients present innumerable asymptomatic liver cysts [33]. However, the data are not sufficient to explain why PKHD1 in ARPKD leads to severe hepatic and renal phenotype, but not in ADPLD; in this regard, more studies are needed [34].
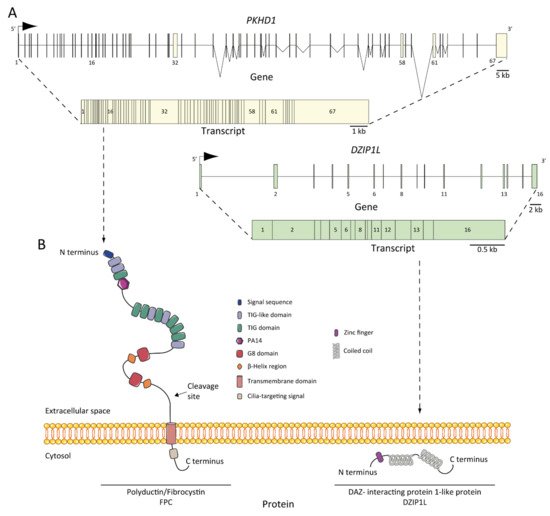
Figure 2. ARPKD genes, transcripts, and proteins: (A) PKHD1 and DZIP1L genes and transcripts. The positions of the exons are illustrated and numbered, and the longest transcript are shown from both: 67 exons for PKHD1 and 16 for DZIP1L; (B) structure of fibrocystin/polyductin (FPC) and DAZ-interacting protein 1-like protein (DZIP1L). Proteins are not to scale.
6. ARPKD Proteins: Structure and Function
The protein product of PKHD1 is fibrocystin/polyductin/FPC (Figure 2B) [21][27], a membrane protein with a long extracellular N-terminus, a single transmembrane domain and a short cytoplasmic C-terminus tail. The extracellular domain contains twelve TIG/IPT domains (Ig-like domains) that have been described in cell surface receptors [35]. In addition, three potential protein kinase A (PKA) phosphorylation sites were identified in the cytoplasmic tail that may be relevant for its function [36]. A PKHD1 homologue was reported, PKHD1L, with an identity of 25% and similarity of 41.5%, which encodes fibrocystin-L, a receptor with inducible T lymphocyte expression, and has not been implicated in PKD [37]. The longest open reading frame (ORF) of FPC is predicted with a length of 4074 amino acids [38]. However, the PKHD1 gene undergoes a complicated splicing pattern and can encode several additional gene products. In the same way, FPC exhibits a highly complex pattern of Notch-like proteolytic processing validated at the in vitro level [39] and in the vivo level using mouse models [40], which make the investigation of PKHD1/FPC particularly difficult.
FPC is a 440 kDa membrane-bound protein that is expressed mainly in the kidney (cortical and medullary ducts), the liver (intra- and extra-hepatic biliary ducts) and the pancreas (pancreatic ducts) [38][41][42]. Two alternative FPC products of ~230 and ~140 kDa were detected and, more importantly, the ~140 kDa product was found in cellular fractions of secreted FPC products [38]. At the subcellular level, FPC is expressed in the primary apical cilia [38][41][43] and the basal body area of cilia [42] in renal epithelial cells and cholangiocytes [44]. Furthermore, FPC is also expressed in the apical membrane and cytoplasm of collecting duct cells [38]. It is controversial whether ARPKD tissues lack FPC expression, some studies support this idea [27][43], but other evidence suggests otherwise [45], suggesting a temporal and spatial expression complexity of FPC splicing variants.
The structure of FPC suggests a possible function of the cell surface receptor, which interacts with extracellular ligand through the N-terminus or transduces intracellular signals to the nucleus through its C-terminus [46]. The cytoplasmic tail can translocate to the nucleus after full-length cleavage [39][47]. However, the intrinsic mechanism of the C-terminus remains unclear, as its deletion in mouse models did not result in renal or hepatic cystic phenotype, suggesting that it is not essential for cyst formation in ARPKD [40].
DZIP1L encodes the DAZ (Deleted in AZoospermia) interacting protein 1-like, a zinc-finger protein with several coiled-coil domains and one C2H2-type zinc finger domain near its N-terminus [25]. The zinc finger protein DZIP1L is involved in primary cilium formation [48], and Lu and colleagues suggest a possible function in the polycystins/PCs (the ADPKD proteins) trafficking [25]. The results highlighted the transition zone of cilia as a new possible vital point to study ARPKD pathogenesis [49].
7. Pathogenesis of ARPKD/Molecular Basis/Disease Mechanism
7.1. ARPKD Rodent Models: Lessons from Animal Models
To date, several animal models have been developed in which they closely resemble human ARPKD (Table 1). Early models of PKD resulted from spontaneous mutations in non-orthologous genes that mimic the recessive trait and phenotype of the disease [50]. The first mouse model reported was the congenital polycystic kidney mouse, or cpk, in 1985 [51]. The development and expression/penetrance of disease and the genetics in this model were extensively studied [50][52]. The cpk model results in a spontaneous mutation in the C57BL/6J (B6) strain, which corresponds with the Cys1 gene [35]. Later, during the 1990s, other models appeared with spontaneous mutations in other loci, including the well-studied pcy mouse [53] that has a mutation in the locus for Nphp3 [54]. Furthermore, BALB/c polycystic kidney (bpk) [55] and the juvenile cystic kidney model (jck) [56] were described and characterized, that had spontaneous mutations in Bicc1 [57] and Nek8 [58] respectively. This was followed by animal models designed by chemical induction, as the juvenile congenital polycystic kidney (jcpk), which was obtained using a chlorambucil mutagenesis program [59]. Interestingly, later studies showed that bpk and jcpk models refined the mutated loci in Bicc1 gene [57][60]. Furthermore, by insertional mutagenesis, the Oak Ridge polycystic kidney or orpk mouse was uncovered from a large-scale insertional mutagenesis program [61][62].
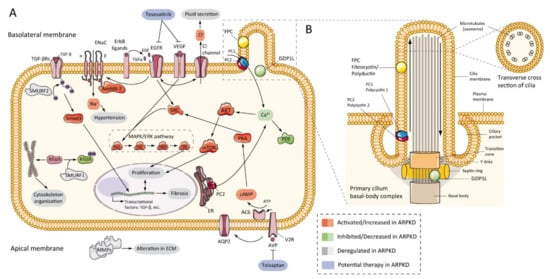
7.2. Abnormalities of EGFR-Axis Expression and Fluid Secretion
The first evidence that the epidermal growth factor receptor (EGFR) axis was altered in PKD was in 1992, by demonstrating that cells from primary cultures of PKD patients increased cyst expansion [63]. Subsequently, in primary cells isolated from ADPKD patients, epidermal growth factor (EGF) stimulated cyst formation [64]. In ARPKD, the first data were obtained from cpk mouse model renal extracts, which showed upregulation of EGF expression [65]. Progressively, other evidence has shown a significant role for EGFR in vitro [66] and murine models [62][67][68], and patients with ARPKD [69][70], where EGFR upregulation was located on the surface of the cystic epithelium. In the same way, abnormal expression of EGF [71][72] and transforming growth factor-alpha (TGFα) [73] have been demonstrated in ARPKD, and several members of EGFR family of receptors (EGFR1, ErbB2, and ErbB4) were found overexpressed in ARPKD rodent models [45][74] (Figure 3A). This overexpression includes increased mRNA, protein, and receptor activity or phosphorylation [75]. Furthermore, evidence from animal models suggests similar abnormalities in hepatic cystogenesis of the EGFR axis [76].Figure 3. ARPKD molecular pathogenesis: (A) diagram representing the proposed up-, down-, or deregulated pathways in ARPKD renal epithelial cell and proposed potential therapies; (B) cartoon representing the localization of ARPKD protein in the cilium of a renal epithelial cell. FPC is located in the primary apical cilia and the basal body area of the cilia, whereas DZIP1L is located in the transition zone of the basal body of the cilium. (FPC—fibrocystin/polyductin; DZIP1L—DAZ interacting zinc finger protein 1 like; PC1—polycystin 1; PC2—polycystin 2; TGF—transforming growth factor; ENaC—epithelial sodium channels; Na+—sodium cation; EGFR—epidermal growth factor receptor; VEGF—vascular endothelial growth factor; Cl−—chlorine anion; SMURF—SMAD specific E3 ubiquitin-protein ligase; Nedd4-2—E3 ubiquitin-protein ligase Nedd4-2; SRC—proto-oncogene tyrosine-protein kinase Src; AKT—RAC-alpha serine/threonine-protein kinase; Ca+—calcium cation; PDE—phosphodiesterase; mTOR—mammalian target of rapamycin; MAPK—mitogen-activated protein kinases; RAF—rapidly accelerated fibrosarcoma; MEK—mitogen-activated protein kinase kinase; ERK—extracellular-signal-regulated kinase; Rhoa—Ras homolog family member A; ER—endoplasmic reticulum; PKA—protein kinase A; cAMP—cyclic adenosine monophosphate (cyclic AMP); ATP—adenosine triphosphate; AC6—adenylate cyclase type 6; AQP2—aquaporin 2; V2R—vasopressin receptor 2; AVP—arginine vasopressin; MMPs—matrix metalloproteinases).
7.3. cAMP and Proliferation
Several studies have shown that adenylyl cyclase adenosine 3′,5′-cyclic monophosphate (cAMP) pathway stimulates cell proliferation in the renal epithelium of ARPKD and ADPKD. Production of cAMP is aberrant in the cyst epithelium, resulting in a large amount of this nucleotide in the cyst fluid [77][78][79][80]. cAMP activates the B-Raf, MEK, and ERK pathways in the cyst epithelium of the kidneys with ADPKD [81][82][83], and ADPKD and ARPKD cells in culture [80]. In the same way, these results were complemented with data showing upregulation at the protein level of MAPK and AKT/mTOR pathways in several rodent models of ARPKD [84][85][86][87][88]. These facts correlated with the reduction of intracellular Ca2+ and the phosphorylation of the SCR protein [89][90][91]. In particular, blocked intracellular Ca2+ elevated AKT and proliferative activity in ARPKD cells in culture [90]. This study opened the opportunity to use the level of intracellular calcium restoration as a therapeutic approach in PKD.
7.4. Other Pathways Involved in ARPKD Physiopathology
Other pathogenic features have been identified in ARPKD, as well as alterations in extracellular matrix (ECM) and metalloproteinase expression (MMPs) [92][93], upregulation of vascular endothelial growth factor (VEGF) and hypoxia-inducible factor-1 alpha (HIF-1α) in Pkhd1 deficient cells [86], upregulation of peroxisome-proliferator-activated receptor-γ (PPAR-γ) in animal models [94][95], or metabolic alterations [96]. In a large and interesting study, Kaimori and colleagues published information about novel functional relationships between FPC and members of the C2-WWW-HECT domain E3 family of ubiquitin ligases. The authors localized FPC in vesicles where Ndfip2 was also present, a ubiquitin ligase interacting protein implicated in trafficking and regulating the Nedd4-2 ubiquitin ligase family and SMURF1 and SMURF2. These data may explain different universal phenotypes in ARPKD and renal and hepatic fibrosis through TGF-β signaling pathways, hypertension through to ENaC mediated sodium reabsorption, and cystogenesis through to RhoA ubiquitination and cytoskeleton organization [97] (Figure 3A). In other studies, tubular morphogenesis in PKD was associated with an abnormality planar cell polarity (PCP) [98]. However, later studies have shown contrary results [99].
7.5. Role of Cilia
Figure 3 shows the ARPKD proteins (zinc finger protein DZIP1L and FPC) are located in the cilia. The cilia are long and microtubular structures emanating from the surface of mammalian cells. The axoneme of primary cilia contains nine peripheral bundles of microtubules (9 + 0 pattern). Pathologies related to a loss of proper cilia function are called ciliopathies, including ARPKD [100]. PC2, also called TRPP2, is a member of the transient receptor channel (TRP) family and is a calcium-permeable non-selective channel [101]. PC2 and PC1 form a receptor-channel complex, that is involved in the calcium pathway and cilia response [102][103][104] (Figure 3B). FPC has been shown to interact with PC2 in primary cilia and regulates PC2 channel activity [105][106][107]. In addition, it has been reported that the C-terminus of FPC physically interacts with the N-terminus of PC2 in vivo and in vitro, and that Pkhd1-deficient cells exhibit dysregulation of PC2 channel activity [105]. However, Wang and colleagues found no differences in PC2 levels in cells with reduced FPC levels [107]. Other data using a novel Pkhd1 mouse model have shown that deletion of the last exon of Pkhd1, the PC2 binding site, and the nuclear localization signal, had no apparent pathologic effects in mice [40]. In addition, the researchers were unable to co-precipitate FPC-PC2 in kidney samples from the transgenic mouse model. These results suggest that the PC2 binding domain of FPC is not essential for the fibrocystin function [40][108].
References
- Guay-Woodford, L.M.; Muecher, G.; Hopkins, S.D.; Avner, E.D.; Germino, G.G.; Guillot, A.P.; Herrin, J.; Holleman, R.; Irons, D.A.; Primack, W.; et al. The severe perinatal form of autosomal recessive polycystic kidney disease maps to chromosome 6p21.1-p12: Implications for genetic counseling. Am. J. Hum. Genet. 1995, 56, 1101–1107.
- Kaariainen, H. Polycystic kidney disease in children: A genetic and epidemiological study of 82 Finnish patients. J. Med. Genet. 1987, 24, 474–481.
- Alzarka, B.; Morizono, H.; Bollman, J.W.; Kim, D.; Guay-Woodford, L.M. Design and implementation of the hepatorenal fibrocystic disease core center clinical database: A centralized resource for characterizing autosomal recessive polycystic kidney disease and other hepatorenal fibrocystic diseases. Front. Pediatr. 2017, 5.
- Bergmann, C. Genetics of Autosomal Recessive Polycystic Kidney Disease and Its Differential Diagnoses. Front. Pediatr. 2018, 5, 1–13.
- Cordido, A.; Besada-Cerecedo, L.; García-González, M.A. The Genetic and Cellular Basis of Autosomal Dominant Polycystic Kidney Disease—A Primer for Clinicians. Front. Pediatr. 2017, 5, 279.
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4, 50.
- Bergmann, C.; Senderek, J.; Schneider, F.; Dornia, C.; Küpper, F.; Eggermann, T.; Rudnik-Schöneborn, S.; Kirfel, J.; Moser, M.; Büttner, R.; et al. PKHD1 Mutations in Families Requesting Prenatal Diagnosis for Autosomal Recessive Polycystic Kidney Disease (ARPKD). Hum. Mutat. 2004, 23, 487–495.
- Guay-Woodford, L.M.; Desmond, R.A. Autosomal recessive polycystic kidney disease: The clinical experience in North America. Pediatrics 2003, 111, 1072–1080.
- Adeva, M.; El-Youssef, M.; Rossetti, S.; Kamath, P.S.; Kubly, V.; Consugar, M.B.; Milliner, D.M.; King, B.F.; Torres, V.E.; Harris, P.C. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine 2006, 85, 1–21.
- Gunay-Aygun, M.; Avner, E.D.; Bacallao, R.L.; Choyke, P.L.; Flynn, J.T.; Germino, G.G.; Guay-Woodford, L.; Harris, P.; Heller, T.; Ingelfinger, J.; et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: Summary statement of a First National Institutes of Health/Office of Rare Diseases conference. J. Pediatr. 2006, 149, 159–164.
- Liebau, M.C. Early clinical management of autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 2021.
- Bergmann, C.; Senderek, J.; Windelen, E.; Küpper, F.; Middeldorf, I.; Schneider, F.; Dornia, C.; Rudnik-Schöneborn, S.; Konrad, M.; Schmitt, C.P.; et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 2005, 67, 829–848.
- Erger, F.; Brüchle, N.O.; Gembruch, U.; Zerres, K. Prenatal ultrasound, genotype, and outcome in a large cohort of prenatally affected patients with autosomal-recessive polycystic kidney disease and other hereditary cystic kidney diseases. Arch. Gynecol. Obstet. 2017, 295, 897–906.
- Burgmaier, K.; Kilian, S.; Bammens, B.; Benzing, T.; Billing, H.; Büscher, A.; Galiano, M.; Grundmann, F.; Klaus, G.; Mekahli, D.; et al. Clinical courses and complications of young adults with Autosomal Recessive Polycystic Kidney Disease (ARPKD). Sci. Rep. 2019, 9.
- Belin, S.; Delco, C.; Parvex, P.; Hanquinet, S.; Fokstuen, S.; De Tejada, B.M.; Eperon, I. Management of delivery of a fetus with autosomal recessive polycystic kidney disease: A case report of abdominal dystocia and review of the literature. J. Med. Case Rep. 2019, 13.
- Fonck, C.; Chauveau, D.; Gagnadoux, M.F.; Pirson, Y.; Grünfeld, J.P. Autosomal recessive polycystic kidney disease. Nephrol. Dial. Transplant. 2001, 16, 1648–1652.
- Rubio San Simón, A.; Carbayo Jiménez, T.; Vara Martín, J.; Alonso Díaz, C.; Espino Hernández, M. Autosomal recessive polycystic kidney disease in the 21st century: Long-term follow up and outcomes. An. Pediatr. 2018, 91, 120–122.
- Burgmaier, K.; Brandt, J.; Shroff, R.; Witters, P.; Weber, L.T.; Dötsch, J.; Schaefer, F.; Mekahli, D.; Liebau, M.C. Gastrostomy tube insertion in pediatric patients with autosomal recessive polycystic kidney disease (ARPKD): Current practice. Front. Pediatr. 2018, 6.
- Guay-Woodford, L.M. Autosomal recessive polycystic kidney disease: The prototype of the hepato-renal fibrocystic diseases. J. Pediatr. Genet. 2014, 3, 89–101.
- Raina, R.; Chakraborty, R.; Sethi, S.K.; Kumar, D.; Gibson, K.; Bergmann, C. Diagnosis and Management of Renal Cystic Disease of the Newborn: Core Curriculum 2021. Am. J. Kidney Dis. 2021.
- Onuchic, L.F.; Furu, L.; Nagasawa, Y.; Hou, X.; Eggermann, T.; Ren, Z.; Bergmann, C.; Senderek, J.; Esquivel, E.; Zeltner, R.; et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 2002, 70, 1305–1317.
- Obeidova, L.; Seeman, T.; Fencl, F.; Blahova, K.; Hojny, J.; Elisakova, V.; Reiterova, J.; Stekrova, J. Results of targeted next-generation sequencing in children with cystic kidney diseases often change the clinical diagnosis. PLoS ONE 2020, 15.
- Bergmann, C. Early and Severe Polycystic Kidney Disease and Related Ciliopathies: An Emerging Field of Interest. Nephron 2019, 141, 50–60.
- Bergmann, C. ARPKD and early manifestations of ADPKD: The original polycystic kidney disease and phenocopies. Pediatr. Nephrol. 2015, 30, 15–30.
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034.
- Zerres, K.; Mücher, G.; Becker, J.; Steinkamm, C.; Rudnik-Schöneborn, S.; Heikkilä, P.; Rapola, J.; Salonen, R.; Germino, G.G.; Onuchic, L.; et al. Prenatal diagnosis of autosomal recessive polycystic kidney disease (ARPKD): Molecular genetics, clinical experience, and fetal morphology. Am. J. Med. Genet. 1998, 76, 137–144.
- Ward, C.J.; Hogan, M.C.; Rossetti, S.; Walker, D.; Sneddon, T.; Wang, X.; Kubly, V.; Cunningham, J.M.; Bacallao, R.; Ishibashi, M.; et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 2002, 30, 259–269.
- Zerres, K.; Rudnik-Schöneborn, S.; Deget, F.; Holtkamp, U.; Brodehl, J.; Geisert, J.; Schärer, K. Autosomal recessive polycystic kidney disease in 115 children: Clinical presentation, course and influence of gender. Arbeitsgemeinschaft für Pädiatrische, Nephrologie. Acta Paediatr. 1996, 85, 437–445.
- Deget, F.; Rudnik-Schöneborn, S.; Zerres, K. Course of autosomal recessive polycystic kidney disease (ARPKD) in siblings: A clinical comparison of 20 sibships. Clin. Genet. 1995, 47, 248–253.
- Consugar, M.B.; Anderson, S.A.; Rossetti, S.; Pankratz, V.S.; Ward, C.J.; Torra, R.; Coto, E.; El-youssef, M.; Kantarci, S.; Utsch, B.; et al. Haplotype Analysis Improves Molecular Diagnostics of Autosomal Recessive Polycystic Kidney Disease. Am. J. Kidney Dis. 2005, 45, 77–87.
- Blyth, H.; Ockenden, B.G. Polycystic disease of kidney and liver presenting in childhood. J. Med. Genet. 1971, 8, 257–284.
- Besse, W.; Dong, K.; Choi, J.; Punia, S.; Fedeles, S.V.; Choi, M.; Gallagher, A.-R.R.; Huang, E.B.; Gulati, A.; Knight, J.; et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Investig. 2017.
- Gunay-aygun, M.; Turkbey, B.I.; Bryant, J.; Daryanani, K.T.; Tuchman, M.; Piwnica-worms, K.; Choyke, P.; Heller, T.; Gahl, W.A. Hepatorenal findings in obligate heterozygotes for autosomal recessive polycystic kidney disease. Mol. Genet. Metab. 2011, 104, 677–681.
- Perugorria, M.J.; Banales, J.M. Genetics: Novel causative genes for polycystic liver disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 391–392.
- Nagasawa, Y.; Matthiesen, S.; Onuchic, L.F.; Hou, X.; Bergmann, C.; Esquivel, E.; Senderek, J.; Ren, Z.; Zeltner, R.; Furu, L.; et al. Identification and Characterization of Pkhd1, the Mouse Orthologue of the Human ARPKD Gene. J. Am. Soc. Nephrol. 2002, 13, 2246–2258.
- Igarashi, P.; Somlo, S. Genetics and Pathogenesis of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2002, 13, 2384–2398.
- Hogan, M.C.; Griffin, M.D.; Rossetti, S.; Torres, V.E.; Ward, C.J.; Harris, P.C. PKHDL1, a homolog of the autosomal recessive polycystic kidney disease gene, encodes a receptor with inducible T lymphocyte expression. Hum. Mol. Genet. 2003, 12, 685–698.
- Menezes, L.F.C.; Cai, Y.; Nagasawa, Y.; Silva, A.M.G.; Watkins, M.L.; Da Silva, A.M.; Somlo, S.; Guay-Woodford, L.M.; Germino, G.G.; Onuchic, L.F. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 2004, 66, 1345–1355.
- Kaimori, J.-Y.; Nagasawa, Y.; Menezes, L.F.; Garcia-Gonzalez, M.A.; Deng, J.; Imai, E.; Onuchic, L.F.; Guay-Woodford, L.M.; Germino, G.G. Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum. Mol. Genet. 2007, 16, 942–956.
- Outeda, P.; Menezes, L.; Hartung, E.A.; Bridges, S.; Zhou, F.; Zhu, X.; Xu, H.; Huang, Q.; Yao, Q.; Qian, F.; et al. A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 2017, 92, 1130–1144.
- Ward, C.J.; Yuan, D.; Masyuk, T.V.; Wang, X.; Punyashthiti, R.; Whelan, S.; Bacallao, R.; Torra, R.; LaRusso, N.F.; Torres, V.E.; et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 2003, 12, 2703–2710.
- Wang, S.; Luo, Y.; Wilson, P.D.; Witman, G.B.; Zhou, J. The Autosomal Recessive Polycystic Kidney Disease Protein Is Localized to Primary Cilia, with Concentration in the Basal Body Area. J. Am. Soc. Nephrol. 2004, 15, 592–602.
- Zhang, M.-Z.; Mai, W.; Li, C.; Cho, S.; Hao, C.; Moeckel, G.; Zhao, R.; Kim, I.; Wang, J.; Xiong, H.; et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 2311–2316.
- Masyuk, T.V.; Huang, B.Q.; Ward, C.J.; Masyuk, A.I.; Yuan, D.; Splinter, P.L.; Punyashthiti, R.; Ritman, E.L.; Torres, V.E.; Harris, P.C.; et al. Defects in Cholangiocyte Fibrocystin Expression and Ciliary Structure in the PCK Rat. Gastroenterology 2003, 125, 1303–1310.
- Sweeney, W.E.; Avner, E.D. Molecular and cellular pathophysiology of autosomal recessive polycystic kidney disease (ARPKD). Cell Tissue Res. 2006, 326, 671–685.
- Wilson, P.D. Polycystic Kidney Disease. N. Engl. J. Med. 2004, 350, 151–164.
- Follit, J.A.; Li, X.; Vucica, Y.; Pazour, G.J. The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J. Cell Biol. 2010, 188, 21–28.
- Glazer, A.M.; Wilkinson, A.W.; Backer, C.B.; Lapan, S.W.; Gutzman, J.H.; Cheeseman, I.M.; Reddien, P.W. The Zn Finger protein Iguana impacts Hedgehog signaling by promoting ciliogenesis. Dev. Biol. 2010, 337, 148–156.
- Hartung, E.A.; Guay-Woodford, L.M. Polycystic kidney disease: DZIP1L defines a new functional zip code for autosomal recessive PKD. Nat. Rev. Nephrol. 2017, 13, 519–520.
- Guay-Woodford, L.M. Murine models of polycystic kidney disease: Molecular and therapeutic insights. Am. J. Physiol. Ren. Physiol. 2003, 285, 1034–1049.
- Fry, J.L.; Koch, W.E.; Jennette, J.C.; McFarland, E.; Fried, F.A.; Mandell, J. A genetically determined murine model of infantile polycystic kidney disease. J. Urol. 1985, 134, 828–833.
- Schieren, G.; Pey, R.; Bach, J.; Hafner, M.; Gretz, N. Murine models of polycystic kidney disease. Nephrol. Dial. Transplant. 1996, 11, 38–45.
- Takahashi, H.; Calvet, J.P.; Dittemore-Hoover, D.; Yoshida, K.; Grantham, J.J.; Gattone, V.H. A hereditary model of slowly progressive polycystic kidney disease in the mouse. J. Am. Soc. Nephrol. 1991, 1, 980–989.
- Woo, D.D.L.; Nguyen, D.K.P.; Khatibi, N.; Olsen, P. Genetic identification of two major modifier loci of polycystic kidney disease progression in pcy mice. J. Clin. Investig. 1997, 100, 1934–1940.
- Nauta, J.; Ozawa, Y.; Sweeney, W.E.; Rutledge, J.C.; Avner, E.D. Renal and biliary abnormalities in a new murine model of autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 1993, 7, 163–172.
- Atala, A.; Freeman, M.R.; Mandell, J.; Beier, D.R. Juvenile cystic kidneys (jck): A new mouse mutation which causes polycystic kidneys. Kidney Int. 1993, 43, 1081–1085.
- Cogswell, C.; Price, S.J.; Hou, X.; Guay-Woodford, L.M.; Flaherty, L.; Bryda, E.C. Positional cloning of jcpk/bpk locus of the mouse. Mamm. Genome 2003, 14, 242–249.
- Liu, S.; Lu, W.; Obara, T.; Kuida, S.; Lehoczky, J.; Dewar, K.; Drummond, I.A.; Beier, D.R. A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 2002, 129, 5839–5846.
- Flaherty, L.; Bryda, E.C.; Collins, D.; Rudofsky, U.; Montgomery, J.C. New mouse model for polycystic kidney disease with both recessive and dominant gene effects. Kidney Int. 1995, 47, 552–558.
- Guay-Woodford, L.M.; Bryda, E.C.; Christine, B.; Lindsey, J.R.; Collier, W.R.; Avner, E.D.; D’eustachio, P.; Flaherty, L. Evidence that two phenotypically distinct mouse PKD mutations, bpk and jcpk, are allelic. Kidney Int. 1996, 50, 1158–1165.
- Moyer, J.H.; Lee-Tischler, M.J.; Kwon, H.Y.; Schrick, J.J.; Avner, E.D.; Sweeney, W.E.; Godfrey, V.L.; Cacheiro, N.L.A.; Wilkinson, J.E.; Woychik, R.P. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science 1994, 264, 1329–1333.
- Sweeney, W.E.; Avner, E.D. Functional activity of epidermal growth factor receptors in autosomal recessive polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 1998, 275, F387–F394.
- Neufeld, T.K.; Douglass, D.; Grant, M.; Ye, M.; Silva, F.; Nadasdy, T.; Grantham, J.J. In vitro formation and expansion of cysts derived from human renal cortex epithelial cells. Kidney Int. 1992, 41, 1222–1236.
- Ye, M.; Grant, M.; Sharma, M.; Elzinga, L.; Swan, S.; Torres, V.E.; Grantham, J.J. Cyst fluid from human autosomal dominant polycystic kidneys promotes cyst formation and expansion by renal epithelial cells in vitro. J. Am. Soc. Nephrol. 1992, 3, 984–994.
- Lakshmanan, J.; Fisher, D.A. An inborn error in epidermal growth factor prohormone metabolism in a mouse model of autosomal recessive polycystic kidney disease. Biochem. Biophys. Res. Commun. 1993, 196, 892–901.
- Pugh, J.L.; Sweeney, W.E.; Avner, E.D. Tyrosine kinase activity of the EGF receptor in murine metanephric organ culture. Kidney Int. 1995, 47, 774–781.
- Dell, K.M.R.; Nemo, R.; Sweeney, W.E.; Avner, E.D. EGF-related growth factors in the pathogenesis of murine ARPKD. Kidney Int. 2004, 65, 2018–2029.
- Richards, W.G.; Sweeney, W.E.; Yoder, B.K.; Wilkinson, J.E.; Woychik, R.P.; Avner, E.D. Epidermal growth factor receptor activity mediates renal cyst formation in polycystic kidney disease. J. Clin. Investig. 1998, 101, 935–939.
- Arbeiter, A.; Büscher, R.; Bonzel, K.E.; Wingen, A.M.; Vester, U.; Wohlschläger, J.; Zerres, K.; Nürnberger, J.; Bergmann, C.; Hoyer, P.F. Nephrectomy in an autosomal recessive polycystic kidney disease (ARPKD) patient with rapid kidney enlargement and increased expression of EGFR. Nephrol. Dial. Transplant. 2008, 23, 3026–3029.
- Rohatgi, R.; Zavilowitz, B.; Vergara, M.; Woda, C.; Kim, P.; Satlin, L.M. Cyst fluid composition in human autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 2005, 20, 552–553.
- Gattone, V.H.; Calvet, J.P. Murine Infantile Polycystic Kidney Disease: A Role for Reduced Renal Epidermal Growth Factor. Am. J. Kidney Dis. 1991, 17, 606–607.
- Nakanishi, K.; Gattone, V.H.; Sweeney, W.E.; Avner, E.D. Renal dysfunction but not cystic change is ameliorated by neonatal epidermal growth factor in bpk mice. Pediatr. Nephrol. 2001, 16, 45–50.
- Lowden, D.A.; Lindemann, G.W.; Merlino, G.; Barash, B.D.; Calvet, J.P.; Gattone, V.H. Renal cysts in transgenic mice expressing transforming gorwth factor-alpha. J. Lab. Clin. Med. 1994, 124, 386–394.
- Zheleznova, N.N.; Wilson, P.D.; Staruschenko, A. Epidermal growth factor-mediated proliferation and sodium transport in normal and PKD epithelial cells. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1301–1313.
- Sweeney, W.E.; Avner, E.D. Pathophysiology of childhood polycystic kidney diseases: New insights into disease-specific therapy. Pediatr. Res. 2014, 75, 148–157.
- Nauta, J.; Sweeney, W.E.; Rutledge, J.C.; Avner, E.D. Biliary epithelial cells from mice with congenital polycystic kidney disease are hyperresponsive to epidermal growth factor. Pediatr. Res. 1995, 37, 755–763.
- Torres, V.E.; Sweeney, W.E.; Wang, X.; Qian, Q.; Harris, P.C.; Frost, P.; Avner, E.D. Epidermal growth factor receptor tyrosine kinase inhibition is not protective in PCK rats. Kidney Int. 2004, 66, 1766–1773.
- Yamaguchi, T.; Nagao, S.; Kasahara, M.; Takahashi, H.; Grantham, J.J. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am. J. Kidney Dis. 1997, 30, 703–709.
- Smith, L.A.; Bukanov, N.O.; Husson, H.; Russo, R.J.; Barry, T.C.; Taylor, A.L.; Beier, D.R.; Ibraghimov-Beskrovnaya, O. Development of polycystic kidney disease in juvenile cystic kidney mice: Insights into pathogenesis, ciliary abnormalities, and common features with human disease. J. Am. Soc. Nephrol. 2006, 17, 2821–2831.
- Belibi, F.A.; Reif, G.; Wallace, D.P.; Yamaguchi, T.; Olsen, L.; Li, H.; Helmkamp, G.M.; Grantham, J.J. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004, 66, 964–973.
- Yamaguchi, T.; Pelling, J.C.; Ramaswamy, N.T.; Eppler, J.W.; Wallace, D.P.; Nagao, S.; Rome, L.A.; Sullivan, L.P.; Grantham, J.J. cAMP stimulates the in vitro proliferation of renal cyst epithelial cells by activating the extracellular signal-regulated kinase pathway. Kidney Int. 2000, 57, 1460–1471.
- Yamaguchi, T.; Nagao, S.; Wallace, D.P.; Belibi, F.A.; Cowley, B.D.; Pelling, J.C.; Grantham, J.J. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003, 63, 1983–1994.
- Yamaguchi, T.; Wallace, D.P.; Magenheimer, B.S.; Hempson, S.J.; Grantham, J.J.; Calvet, J.P. Calcium restriction allows cAMP activation of the B-Raf/ERK pathway, switching cells to a cAMP-dependent growth-stimulated phenotype. J. Biol. Chem. 2004, 279, 40419–40430.
- Wang, X. Effectiveness of Vasopressin V2 Receptor Antagonists OPC-31260 and OPC-41061 on Polycystic Kidney Disease Development in the PCK Rat. J. Am. Soc. Nephrol. 2005, 16, 846–851.
- Wang, X.; Wu, Y.; Ward, C.J.; Harris, P.C.; Torres, V.E. Vasopressin directly regulates cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 102–108.
- Zheng, R.; Wang, L.; Fan, J.; Zhou, Q. Inhibition of PKHD1 may cause S-phase entry via mTOR signaling pathway. Cell Biol. Int. 2009, 33, 926–933.
- Fischer, D.C.; Jacoby, U.; Pape, L.; Ward, C.J.; Kuwertz-Broeking, E.; Renken, C.; Nizze, H.; Querfeld, U.; Rudolph, B.; Mueller-Wiefel, D.E.; et al. Activation of the AKTmTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol. Dial. Transplant. 2009, 24, 1819–1827.
- Ren, X.S.; Sato, Y.; Harada, K.; Sasaki, M.; Furubo, S.; Song, J.Y.; Nakanuma, Y. Activation of the PI3K/mTOR Pathway Is Involved in Cystic Proliferation of Cholangiocytes of the PCK Rat. PLoS ONE 2014, 9, e87660.
- Hovater, M.B.; Olteanu, D.; Hanson, E.L.; Cheng, N.-L.; Siroky, B.; Fintha, A.; Komlosi, P.; Liu, W.; Satlin, L.M.; Darwin Bell, P.; et al. Loss of apical monocilia on collecting duct principal cells impairs ATP secretion across the apical cell surface and ATP-dependent and flow-induced calcium signals. Purinergic Signal. 2007.
- Yamaguchi, T.; Hempson, S.J.; Reif, G.A.; Hedge, A.M.; Wallace, D.P. Calcium restores a normal proliferation phenotype in human polycystic kidney disease epithelial cells. J. Am. Soc. Nephrol. 2006, 17, 178–187.
- Sweeney, W.E.; von Vigier, R.O.; Frost, P.; Avner, E.D. Src inhibition ameliorates polycystic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 1331–1341.
- Liu, B.; Li, C.; Liu, Z.; Dai, Z.; Tao, Y. Increasing extracellular matrix collagen level and MMP activity induces cyst development in polycystic kidney disease. BMC Nephrol. 2012, 13, 1–8.
- Urribarri, A.D.; Munoz-Garrido, P.; Perugorria, M.J.; Erice, O.; Merino-Azpitarte, M.; Arbelaiz, A.; Lozano, E.; Hijona, E.; Jiménez-Agüero, R.; Fernandez-Barrena, M.G.; et al. Inhibition of metalloprotease hyperactivity in cystic cholangiocytes halts the development of polycystic liver diseases. Gut 2014, 63, 1658–1667.
- Dai, B.; Liu, Y.; Mei, C.; Fu, L.; Xiong, X.; Zhang, Y.; Shen, X.; Hua, Z. Rosiglitazone attenuates development of polycystic kidney disease and prolongs survival in Han:SPRD rats. Clin. Sci. 2010, 119, 323–333.
- Yoshihara, D.; Kugita, M.; Yamaguchi, T.; Aukema, H.M.; Kurahashi, H.; Morita, M.; Hiki, Y.; Calvet, J.P.; Wallace, D.P.; Toyohara, T.; et al. Global gene expression profiling in PPAR-γ agonist-treated kidneys in an orthologous rat model of human autosomal recessive polycystic kidney disease. PPAR Res. 2012.
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wüthrich, R.P. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLoS ONE 2016, 11.
- Kaimori, J.; Lin, C.-C.; Outeda, P.; Garcia-Gonzalez, M.A.; Menezes, L.F.; Hartung, E.A.; Li, A.; Wu, G.; Fujita, H.; Sato, Y.; et al. NEDD4-family E3 ligase dysfunction due to PKHD1/Pkhd1 defects suggests a mechanistic model for ARPKD pathobiology. Sci. Rep. 2017, 7, 7733.
- Fischer, E.; Legue, E.; Doyen, A.; Nato, F.; Nicolas, J.F.; Torres, V.; Yaniv, M.; Pontoglio, M. Defective planar cell polarity in polycystic kidney disease. Nat. Genet. 2006, 38, 21–23.
- Nishio, S.; Tian, X.; Gallagher, A.R.; Yu, Z.; Patel, V.; Igarashi, P.; Somlo, S. Loss of Oriented Cell Division Does not Initiate Cyst Formation. J. Am. Soc. Nephrol. 2010, 21, 295–302.
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543.
- Busch, T.; Köttgen, M.; Hofherr, A. TRPP2 ion channels: Critical regulators of organ morphogenesis in health and disease. Cell Calcium 2017, 66, 25–32.
- Hanaoka, K.; Qian, F.; Boletta, A.; Bhunia, A.K.; Piontek, K.; Tsiokas, L.; Sukhatme, V.P.; Guggino, W.B.; Germino, G.G. Co-assembly of polycystin-1 and -2 produces unique cation-permeable currents. Nature 2000, 408, 990–994.
- Nauli, S.M.; Alenghat, F.J.; Luo, Y.; Williams, E.; Vassilev, P.; Li, X.; Elia, A.E.H.; Lu, W.; Brown, E.M.; Quinn, S.J.; et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 2003, 33, 129–137.
- Wang, Z.; Ng, C.; Liu, X.; Wang, Y.; Li, B.; Kashyap, P.; Chaudhry, H.A.; Castro, A.; Kalontar, E.M.; Ilyayev, L.; et al. The ion channel function of polycystin-1 in the polycystin-1/polycystin-2 complex. EMBO Rep. 2019, 1–18.
- Kim, I.; Fu, Y.; Hui, K.; Moeckel, G.; Mai, W.; Li, C.; Liang, D.; Zhao, P.; Ma, J.; Chen, X.-Z.; et al. Fibrocystin/Polyductin Modulates Renal Tubular Formation by Regulating Polycystin-2 Expression and Function. J. Am. Soc. Nephrol. 2008, 19, 455–468.
- Wu, Y.; Dai, X.-Q.Q.; Li, Q.; Chen, C.X.; Mai, W.; Hussain, Z.; Long, W.; Montalbetti, N.; Li, G.; Glynne, R.; et al. Kinesin-2 mediates physical and functional interactions between polycystin-2 and fibrocystin. Hum. Mol. Genet. 2006, 15, 3280–3292.
- Wang, S.; Zhang, J.; Nauli, S.M.; Li, X.; Starremans, P.G.; Luo, Y.; Roberts, K.A.; Zhou, J. Fibrocystin/Polyductin, Found in the Same Protein Complex with Polycystin-2, Regulates Calcium Responses in Kidney Epithelia Fibrocystin/Polyductin, Found in the Same Protein Complex with Polycystin-2, Regulates Calcium Responses in Kidney Epithel. Mol. Cell. Biol. 2007, 27, 3241–3252.
- Allison, S.J. Polycystic kidney disease: FPC in ARPKD. Nat. Rev. Nephrol. 2017, 13, 597.
More