The SARS-CoV-2 pandemic introduced the world to a new type of vaccine based on mRNA encapsulated in lipid nanoparticles (LNPs). Instead of delivering antigenic proteins directly, an mRNA-based vaccine relies on the host’s cells to manufacture protein immunogens which, in turn, are targets for antibody and cytotoxic T cell responses. mRNA-based vaccines have been the subject of research for over three decades as a platform to protect against or treat a variety of cancers, amyloidosis and infectious diseases. In this review, we discuss mRNA-based approaches for the generation of prophylactic and therapeutic vaccines to HIV. We examine the special immunological hurdles for a vaccine to elicit broadly neutralizing antibodies and effective T cell responses to HIV. Lastly, we outline an mRNA-based HIV vaccination strategy based on the immunobiology of broadly neutralizing antibody development.
- HIV
- vaccine
- messenger RNA
1. Progress in mRNA Technology for HIV Vaccines
1.1. Non-Amplifying mRNA Vaccines
1.2. Self-Amplifying RNA Vaccines
1.3. Nucleoside Modification of mRNA
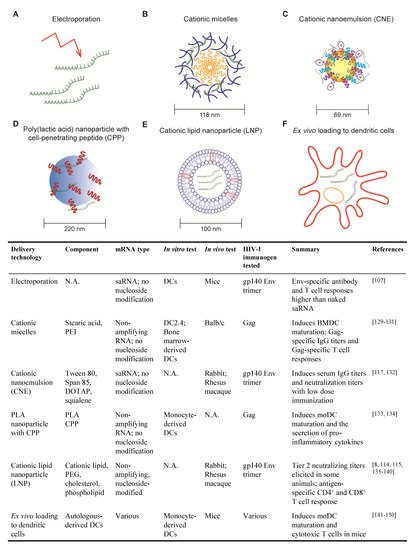
2. Progress in mRNA Delivery Strategies for HIV Vaccines
2.1. Electroporation
2.2. Cationic Micelles
2.3. Cationic Nanoemulsion
2.4. Poly (lactic acid) Nanoparticle with Cell-Penetrating Peptides
2.5. Cationic Lipid Nanoparticle
2.6. Ex Vivo Loading of Dendritic Cell
References
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081.
- Cu, Y.; Broderick, K.E.; Banerjee, K.; Hickman, J.; Otten, G.; Barnett, S.; Kichaev, G.; Sardesai, N.Y.; Ulmer, J.B.; Geall, A. Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ. Vaccines (Basel) 2013, 1, 367–383.
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468.
- D’Haese, S.; Lacroix, C.; Garcia, F.; Plana, M.; Ruta, S.; Vanham, G.; Verrier, B.; Aerts, J.L. Off the beaten path: Novel mRNA-nanoformulations for therapeutic vaccination against HIV. J. Control. Release 2020.
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279.
- Pardi, N.; Weissman, D. Nucleoside Modified mRNA Vaccines for Infectious Diseases. Methods Mol. Biol. 2017, 1499, 109–121.
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2020.
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216.
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magne, R.; Gomard, E.; Guillet, J.G.; Levy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722.
- Alameh, M.G.; Weissman, D.; Pardi, N. Messenger RNA-Based Vaccines Against Infectious Diseases. Curr. Top. Microbiol. Immunol. 2020.
- Anderson, E.J.; Rouphael, N.G.; Widge, A.T.; Jackson, L.A.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Safety and Immunogenicity of SARS-CoV-2 mRNA-1273 Vaccine in Older Adults. N. Engl. J. Med. 2020.
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based Covid-19 Vaccine Candidates. N. Engl. J. Med. 2020.
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The new revolution in nucleic acid vaccines. Semin. Immunol. 2013, 25, 152–159.
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J. Infect. Dis. 2015, 211, 947–955.
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609.
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20.
- Kariko, K.; Buckstein, M.; Ni, H.P.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175.
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 23, 165–175.
- Anderson, B.R.; Muramatsu, H.; Jha, B.K.; Silverman, R.H.; Weissman, D.; Kariko, K. Nucleoside modifications in RNA limit activation of 2′-5′-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011, 39, 9329–9338.
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three decades of messenger RNA vaccine development. Nano Today 2019, 28.
- Kauffman, K.J.; Mir, F.F.; Jhunjhunwala, S.; Kaczmarek, J.C.; Hurtado, J.E.; Yang, J.H.; Webber, M.J.; Kowalski, P.S.; Heartlein, M.W.; DeRosa, F.; et al. Efficacy and immunogenicity of unmodified and pseudouridine-modified mRNA delivered systemically with lipid nanoparticles in vivo. Biomaterials 2016, 109, 78–87.
- Pollard, C.; Rejman, J.; De Haes, W.; Verrier, B.; Van Gulck, E.; Naessens, T.; De Smedt, S.; Bogaert, P.; Grooten, J.; Vanham, G.; et al. Type I IFN counteracts the induction of antigen-specific immune responses by lipid-based delivery of mRNA vaccines. Mol. Ther. 2013, 21, 251–259.
- Kariko, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840.
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Kariko, K. Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic. Acids Res. 2010, 38, 5884–5892.
- Kariko, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic. Acids Res. 2011, 39, e142.
- Weissman, D.; Pardi, N.; Muramatsu, H.; Kariko, K. HPLC purification of in vitro transcribed long RNA. Methods Mol. Biol. 2013, 969, 43–54.
- Zeng, Q.; Jiang, H.; Wang, T.; Zhang, Z.; Gong, T.; Sun, X. Cationic micelle delivery of Trp2 peptide for efficient lymphatic draining and enhanced cytotoxic T-lymphocyte responses. J. Control Release 2015, 200, 1–12.
- Zhao, M.; Li, M.; Zhang, Z.; Gong, T.; Sun, X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA. Drug Deliv. 2016, 23, 2596–2607.
- Li, M.; Zhao, M.; Fu, Y.; Li, Y.; Gong, T.; Zhang, Z.; Sun, X. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra- and paracellular pathways. J. Control Release 2016, 228, 9–19.
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129.
- Tyler, B.; Gullotti, D.; Mangraviti, A.; Utsuki, T.; Brem, H. Polylactic acid (PLA) controlled delivery carriers for biomedical applications. Adv. Drug Deliv. Rev. 2016, 107, 163–175.
- Coolen, A.L.; Lacroix, C.; Mercier-Gouy, P.; Delaune, E.; Monge, C.; Exposito, J.Y.; Verrier, B. Poly(lactic acid) nanoparticles and cell-penetrating peptide potentiate mRNA-based vaccine expression in dendritic cells triggering their activation. Biomaterials 2019, 195, 23–37.
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control Release 2015, 217, 345–351.
- Morrissey, D.V.; Lockridge, J.A.; Shaw, L.; Blanchard, K.; Jensen, K.; Breen, W.; Hartsough, K.; Machemer, L.; Radka, S.; Jadhav, V.; et al. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005, 23, 1002–1007.
- Corbett, K.S.; Flynn, B.; Foulds, K.E.; Francica, J.R.; Boyoglu-Barnum, S.; Werner, A.P.; Flach, B.; O’Connell, S.; Bock, K.W.; Minai, M.; et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N. Engl. J. Med. 2020, 383, 1544–1555.
- Pardi, N.; LaBranche, C.C.; Ferrari, G.; Cain, D.W.; Tombacz, I.; Parks, R.J.; Muramatsu, H.; Mui, B.L.; Tam, Y.K.; Kariko, K.; et al. Characterization of HIV-1 Nucleoside-Modified mRNA Vaccines in Rabbits and Rhesus Macaques. Mol. Ther. Nucleic Acids 2019, 15, 36–47.
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588.
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630.
- Weissman, D.; Ni, H.; Scales, D.; Dude, A.; Capodici, J.; McGibney, K.; Abdool, A.; Isaacs, S.N.; Cannon, G.; Karikó, K. HIV gag mRNA transfection of dendritic cells (DC) delivers encoded antigen to MHC class I and II molecules, causes DC maturation, and induces a potent human in vitro primary immune response. J. Immunol. 2000, 165, 4710–4717.
- Saeboe-Larssen, S.; Fossberg, E.; Gaudernack, G. mRNA-based electrotransfection of human dendritic cells and induction of cytotoxic T lymphocyte responses against the telomerase catalytic subunit (hTERT). J. Immunol. Methods 2002, 259, 191–203.
- Van Tendeloo, V.F.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: Superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98, 49–56.
- Routy, J.P.; Boulassel, M.R.; Yassine-Diab, B.; Nicolette, C.; Healey, D.; Jain, R.; Landry, C.; Yegorov, O.; Tcherepanova, I.; Monesmith, T.; et al. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin. Immunol. 2010, 134, 140–147.
- Allard, S.D.; De Keersmaecker, B.; de Goede, A.L.; Verschuren, E.J.; Koetsveld, J.; Reedijk, M.L.; Wylock, C.; De Bel, A.V.; Vandeloo, J.; Pistoor, F.; et al. A phase I/IIa immunotherapy trial of HIV-1-infected patients with Tat, Rev and Nef expressing dendritic cells followed by treatment interruption. Clin. Immunol. 2012, 142, 252–268.
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E.; et al. Immunization of HIV-1-Infected Persons With Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J. Acquir. Immun. Defic. Syndr. 2016, 71, 246–253.
- Jacobson, J.M.; Routy, J.P.; Welles, S.; DeBenedette, M.; Tcherepanova, I.; Angel, J.B.; Asmuth, D.M.; Stein, D.K.; Baril, J.G.; McKellar, M.; et al. Dendritic Cell Immunotherapy for HIV-1 Infection Using Autologous HIV-1 RNA: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Jaids-J. Acq. Imm. Def. 2016, 72, 31–38.
- Gay, C.L.; DeBenedette, M.A.; Tcherepanova, I.Y.; Gamble, A.; Lewis, W.E.; Cope, A.B.; Kuruc, J.D.; McGee, K.S.; Kearney, M.F.; Coffin, J.M.; et al. Immunogenicity of AGS-004 Dendritic Cell Therapy in Patients Treated During Acute HIV Infection. Aids Res. Hum. Retrovir. 2018, 34, 111–122.
- Guardo, A.C.; Joe, P.T.; Miralles, L.; Bargalló, M.E.; Mothe, B.; Krasniqi, A.; Heirman, C.; García, F.; Thielemans, K.; Brander, C.; et al. Preclinical evaluation of an mRNA HIV vaccine combining rationally selected antigenic sequences and adjuvant signals (HTI-TriMix). Aids 2017, 31, 321–332.
- Jong, W.; Leal, L.; Buyze, J.; Pannus, P.; Guardo, A.; Salgado, M.; Mothe, B.; Molto, J.; Moron-Lopez, S.; Gálvez, C.; et al. Therapeutic Vaccine in Chronically HIV-1-Infected Patients: A Randomized, Double-Blind, Placebo-Controlled Phase IIa Trial with HTI-TriMix. Vaccines 2019, 7, 209.