Bladder cancer (BC) represents a clinical, social, and economic challenge due to tumor-intrinsic characteristics, limitations of diagnostic techniques and a lack of personalized treatments. In the last decade, the use of liquid biopsy has grown as a non-invasive approach to characterize tumors. Moreover, the emergence of omics has increased our knowledge of cancer biology and identified critical BC biomarkers. The rewiring between epigenetics and metabolism has been closely linked to tumor phenotype. Chromatin remodelers interact with each other to control gene silencing in BC, but also with stress-inducible factors or oncogenic signaling cascades to regulate metabolic reprogramming towards glycolysis, the pentose phosphate pathway, and lipogenesis. Concurrently, one-carbon metabolism supplies methyl groups to histone and DNA methyltransferases, leading to the hypermethylation and silencing of suppressor genes in BC. Conversely, α-KG and acetyl-CoA enhance the activity of histone demethylases and acetyl transferases, increasing gene expression, while succinate and fumarate have an inhibitory role.
- bladder cancer
- metabolic pathways
- metabolism
- epigenetics
- biomarkers
1. Introduction
2. Metabolic Rewiring Controls the Epigenome in BC
2.1. Metabolites and DNA/Histone Methylation Processes
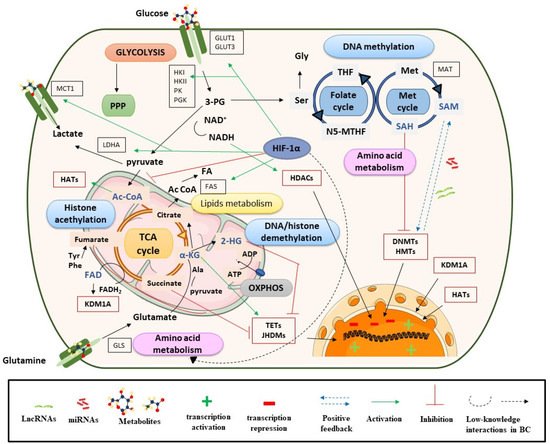
2.2. Metabolites, Histone Acetylation Processes and Sirtuins
2.3. Role of Metabolites in the Nucleus
References
- Burger, M.; Catto, J.W.F.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and risk factors of urothelial bladder cancer. Eur. Urol. 2013, 63, 234–241.
- Kaseb, H.; Aeddula, N.R. Bladder Cancer. En: StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA. Available online: (accessed on 20 August 2020).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca. Cancer J. Clin. 2018, 68, 394–424.
- Anastasiadis, A.; de Reijke, T.M. Best practice in the treatment of nonmuscle invasive bladder cancer. Adv. Urol. 2012, 4, 13–32.
- Knowles, M.A.; Hurst, C.D. Molecular biology of bladder cancer: New insights into pathogenesis and clinical diversity. Nat. Publ. Gr. 2015, 15, 25–41.
- Ghatalia, P.; Zibelman, M.; Geynisman, D.M.; Plimack, E. Approved checkpoint inhibitors in bladder cancer: Which drug should be used when? Adv. Med. Oncol. 2018, 10.
- Rosser, C.J.; Urquidi, V.; Goodison, S. Urinary biomarkers of bladder cancer: An update and future perspectives. Biomark. Med. 2013, 7, 779–790.
- Cauberg Evelyne, C.; de Reijke, T.; de la Rosette, J.M.C. Emerging optical techniques in advanced cystoscopy for bladder cancer diagnosis: A review of the current literature. Indian J. Urol. 2011, 27, 245.
- Babjuk, M.; Böhle, A.; Burger, M.; Capoun, O.; Cohen, D.; Compérat, E.M.; Hernández, V.; Kaasinen, E.; Palou, J.; Rouprêt, M.; et al. EAU Guidelines on Non–Muscle-invasive Urothelial Carcinoma of the Bladder: Update 2016. Eur. Urol. 2017, 71, 447–461.
- Svatek, R.S.; Hollenbeck, B.K.; Holmäng, S.; Lee, R.; Kim, S.P.; Stenzl, A.; Lotan, Y. The economics of bladder cancer: Costs and considerations of caring for this disease. Eur. Urol. 2014, 66, 253–262.
- Leal, J.; Luengo-Fernandez, R.; Sullivan, R.; Witjes, J.A. Economic Burden of Bladder Cancer across the European Union. Eur. Urol. 2016, 69, 438–447.
- Sylvester, R.J.; Van Der Meijden, A.P.M.; Oosterlinck, W.; Witjes, J.A.; Bouffioux, C.; Denis, L.; Newling, D.W.W.; Kurth, K. Predicting recurrence and progression in individual patients with stage Ta T1 bladder cancer using EORTC risk tables: A combined analysis of 2596 patients from seven EORTC trials. Eur. Urol. 2006, 49, 466–475.
- Lodewijk, I.; Dueñas, M.; Rubio, C.; Munera-Maravilla, E.; Segovia, C.; Bernardini, A.; Teijeira, A.; Paramio, J.M.; Suárez-Cabrera, C. Liquid Biopsy Biomarkers in Bladder Cancer: A Current Need for Patient Diagnosis and Monitoring. Int. J. Mol. Sci. 2019, 19, 2514.
- Loras, A.; Su, C.; Mart, M.C. Integrative Metabolomic and Transcriptomic Analysis. Cancers 2019, 11, 686.
- De Oliveira, M.C.; Caires, H.R.; Oliveira, M.J.; Fraga, A.; Vasconcelos, M.H.; Ribeiro, R. Urinary Biomarkers in Bladder Cancer: Where Do We Stand and Potential Role of Extracellular Vesicles. Cancers 2020, 12, 1400.
- Armitage, E.G.; Ciborowski, M. Applications of Metabolomics in Cancer Studies. Adv. Exp. Med. Biol. 2017, 965, 209–234.
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47.
- Holmes, E.; Wilson, I.D.; Nicholson, J.K. Metabolic phenotyping in health and disease. Cell 2008, 134, 714–717.
- Smolinska, A.; Blanchet, L.; Buydens, L.M.; Wijmenga, S.S. NMR and pattern recognition methods in metabolomics: From data acquisition to biomarker discovery: A review. Anal. Chim. Acta 2012, 750, 82–97.
- Crispo, F.; Condelli, V.; Lepore, S.; Notarangelo, T.; Sgambato, A.; Esposito, F.; Maddalena, F.; Landriscina, M. Metabolic Dysregulations and Epigenetics: A Bidirectional Interplay that Drives Tumor Progression. Cells 2019, 8, 798.
- Wong, C.C.; Qian, Y.; Yu, J. Interplay between epigenetics and metabolism in oncogenesis: Mechanisms and therapeutic approaches. Oncogene 2017, 36, 3359–3374.
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380.
- Fouad, Y.A.; Aanei, C. Revisiting the hallmarks of cancer. Am. J. Cancer Res. 2017, 7, 1016–1036.
- Frontiers-Metabolism and Transcription in Cancer: Merging Two Classic Tales-Cell and Developmental Biology [Internet]. Available online: (accessed on 20 August 2020).
- Putluri, N.; Shojaie, A.; Vasu, V.T.; Vareed, S.K.; Nalluri, S.; Putluri, V.; Thangjam, G.S.; Panzitt, K.; Tallman, C.T.; Butler, C.; et al. Metabolomic profiling reveals potential markers and bioprocesses altered in bladder cancer progression. Cancer Res. 2011, 71, 7376–7386.
- Wittmann, B.M.; Stirdivant, S.M.; Mitchell, M.W.; Wulff, J.E.; McDunn, J.E.; Li, Z.; Dennis-Barrie, A.; Neri, B.P.; Milburn, M.V.; Lotan, Y.; et al. Bladder cancer biomarker discovery using global metabolomic profiling of urine. PLoS ONE 2014, 9, e115870.
- Miranda-Gonçalves, V.; Lameirinhas, A.; Henrique, R.; Jerónimo, C. Metabolism and epigenetic interplay in cancer: Regulation and putative therapeutic targets. Front. Genet. 2018, 9, 427.
- Jin, X.; Yun, S.J.; Jeong, P.; Kim, I.Y.; Kim, W.-J.; Park, S. Diagnosis of bladder cancer and prediction of survival by urinary metabolomics. Oncotarget 2014, 5, 1635–1645.
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750.
- Knaap, J.A.; van der Verrijzer, C.P. Undercover: Gene control by metabolites and metabolic enzymes. Genes Dev. 11 Enero 2016, 30, 2345–2369.
- Martínez-reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control. Nat. Commun. 2020, 1–11.
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309.
- Zdzisin, B. Alpha-Ketoglutarate as a Molecule with Pleiotropic Activity: Well-Known and Novel Possibilities of Therapeutic Use. Arch. Immunol. Exp. 2017, 21–36.
- Jun, J.C.; Rathore, A.; Younas, H.; Gilkes, D.; Polotsky, V.Y. Hypoxia-Inducible Factors and Cancer. Curr. Sleep Med. Rep. 2017, 3, 1–10.
- Theodoropoulos, V.E.; Lazaris, A.C.; Sofras, F.; Gerzelis, I.; Tsoukala, V.; Ghikonti, I.; Manikas, K.; Kastriotis, I. Hypoxia-inducible factor 1 alpha expression correlates with angiogenesis and unfavorable prognosis in bladder cancer. Eur. Urol. 2004, 46, 200–208.
- Lew, C.R.; Guin, S.; Theodorescu, D. Targeting glycogen metabolism in bladder cancer. Nat. Rev. Urol. 2015, 12, 383–391.
- Miao, P.; Sheng, S.; Sun, X.; Liu, J.; Huang, G. Lactate dehydrogenase A in cancer: A promising target for diagnosis and therapy. IUBMB Life 2013, 65, 904–910.
- Zhang, G.; Zhang, Y.; Dong, D.; Wang, F.; Ma, X.; Guan, F. MCT1 regulates aggressive and metabolic phenotypes in bladder cancer. J. Cancer 2018, 9.
- Conde, V.R.; Oliveira, P.F.; Nunes, A.R.; Rocha, C.S.; Ramalhosa, E.; Pereira, J.A.; Alves, M.G.; Silva, B.M. The progression from a lower to a higher invasive stage of bladder cancer is associated with severe alterations in glucose and pyruvate metabolism. Exp. Cell Res. 2015, 335, 91–98.
- DeBerardinis, R.J.; Chandel, N.S. We need to talk about the Warburg effect. Nat. Metab. 2020, 2, 127–129.
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214.
- Zhang, J.; Wang, L.; Mao, S.; Liu, M.; Zhang, W.; Zhang, Z.; Guo, Y.; Huang, B.; Yan, Y.; Huang, Y.; et al. MiR-1-3p contributes to cell proliferation and invasion by targeting glutaminase in bladder cancer cells. Cell. Physiol. Biochem. 2018, 51, 513–527.
- Zhou, Q.; Zhan, H.; Lin, F.; Liu, Y.; Yang, K.; Gao, Q.; Ding, M.; Liu, Y.; Huang, W.; Cai, Z. LincRNA-p21 suppresses glutamine catabolism and bladder cancer cell growth through inhibiting glutaminase expression. Biosci. Rep. 2019, 29.
- Zhou, Y.; Song, R.; Zhang, Z.; Lu, X.; Zeng, Z.; Hu, C.; Liu, X.; Li, Y.; Hou, J.; Sun, Y.; et al. The development of plasma pseudotargeted GC-MS metabolic profiling and its application in bladder cancer. Anal. Bioanal. Chem. 2016, 408, 6741–6749.
- Khatami, F.; Aghamir, S.M.K.; Tavangar, S.M. Oncometabolites: A new insight for oncology. Mol. Genet. Genom. Med. 2019, 7.
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181.
- Xiao, M.; Yang, H.; Xu, W.; Ma, S.; Lin, H.; Zhu, H.; Liu, L.; Liu, Y.; Yang, C.; Xu, Y.; et al. Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012, 26, 1326–1338.
- Campbell, S.L.; Wellen, K.E. Metabolic Signaling to the Nucleus in Cancer. Mol. Cell. 2018, 71, 398–408.
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interferon. Cytokine Res. 2016, 36, 226–237.
- Sun, Y.; Cheng, M.K.; Griffiths, T.R.; Mellon, J.K.; Kai, B.; Kriajevska, M.; Manson, M.M. Inhibition of STAT signalling in bladder cancer by diindolylmethane: Relevance to cell adhesion, migration and proliferation. Curr. Cancer Drug Targets 2013, 13, 57–68.
- Hindupur, S.V.; Schmid, S.C.; Koch, J.A.; Youssef, A.; Baur, E.M.; Wang, D.; Horn, T.; Slotta-Huspenina, J.; Gschwend, J.E.; Holm, P.S.; et al. STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy. Int. J. Mol. Sci. 2020, 21, 1106.
- Bernstein, B.E.; Meissner, A.; Lander, E.S. The Mammalian Epigenome. Cell 2007, 128, 669–681.
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3154–3158.
- Zhu, Q.; Hong, B.; Zhang, L.; Wang, J. Pyruvate kinase M2 inhibits the progression of bladder cancer by targeting MAKP pathway. J. Cancer Res. 2018, 14, S616–S621.
- Wang, X.; Zhang, F.; Wu, X.R. Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci. Rep. 2017, 7, 1–13.
- Su, Q.; Tao, T.; Tang, L.; Deng, J.; Darko, K.O.; Zhou, S.; Peng, M.; He, S.; Zeng, Q.; Chen, A.F.; et al. Down-regulation of PKM2 enhances anticancer efficiency of THP on bladder cancer. J. Cell. Mol. Med. 2018, 2774–2790.
- Huang, C.; Huang, Z.; Bai, P.; Luo, G.; Zhao, X.; Wang, X. Expression of pyruvate kinase M2 in human bladder cancer and its correlation with clinical parameters and prognosis. Oncol. Targets 2018, 11, 2075–2082.
- Boukouris, A.E.; Zervopoulos, S.D.; Michelakis, E.D. Metabolic Enzymes Moonlighting in the Nucleus: Metabolic Regulation of Gene Transcription. Trends Biochem. Sci. 2016, 41, 712–730.