Despite its abundance in the environment, iron is poorly bioavailable and subject to strict conservation and internal recycling by most organisms. In vertebrates, the stability of iron concentration in plasma and extracellular fluid, and the total body iron content are maintained by the interaction of the iron-regulatory peptide hormone hepcidin with its receptor and cellular iron exporter ferroportin (SLC40a1). Ferroportin exports iron from duodenal enterocytes that absorb dietary iron, from iron-recycling macrophages in the spleen and the liver, and from iron-storing hepatocytes. Hepcidin blocks iron export through ferroportin, causing hypoferremia. During iron deficiency or after hemorrhage, hepcidin decreases to allow iron delivery to plasma through ferroportin, thus promoting compensatory erythropoiesis. As a host defense mediator, hepcidin increases in response to infection and inflammation, blocking iron delivery through ferroportin to blood plasma, thus limiting iron availability to invading microbes. Genetic diseases that decrease hepcidin synthesis or disrupt hepcidin binding to ferroportin cause the iron overload disorder hereditary hemochromatosis.
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. The Roles of Iron
Iron is an essential trace element for nearly all living organisms. Even though iron is one of the most abundant elements in the Earth’s crust, ferric iron, an oxidized form common in the oxygen-rich environment on the surface of the Earth, is poorly soluble and difficult to access by most life forms. Accordingly, biological organisms have evolved mechanisms that conserve iron and recycle it internally. In adult humans, total body iron content is approximately 3–4 g, whereas normal daily losses are only 1–2 mg. To remain in iron balance, healthy humans must absorb a similar amount of iron from their diets.
The ability of iron to donate or accept an electron in cellular and extracellular environments is what makes it a versatile catalytic component of many enzymes involved in energy-producing bioreactions and critical biosynthetic pathways, as well as in enzymes that generate reactive oxygen species for host defense. Iron also coordinates oxygen in hemoglobin and myoglobin, molecules involved in oxygen transport and its cellular storage. In biological systems, iron carries out its function in association with three common types of moieties: iron coordinated by protein side chains, iron complexed within the porphyrin ring of heme, and iron within iron-sulfur clusters. Outside of these controlled chemical environments, iron displays promiscuous reactivity that can damage cells and tissues.
2. Iron Compartments and Flows
Biological organisms closely regulate intracellular and extracellular iron concentrations, navigating between the twin threats of inadequate iron supply that would limit critical functions, and uncontrolled iron excess that could be toxic to the organism. Systemic iron homeostasis, best understood in humans and laboratory rodents, is maintained by regulating intestinal iron absorption, the concentration of iron in blood plasma and extracellular fluid, the distribution of iron among organs and tissues, and the amount of iron kept in stores. Iron dyshomeostasis can manifest as total body iron deficit (iron deficiency) or excess (iron overload), as well as iron maldistribution among tissues in which individual tissues or organs may become iron-deficient or iron-overloaded. Such iron disorders may be caused by genetic lesions that directly impair iron regulation, or conditions that impact iron regulation indirectly.
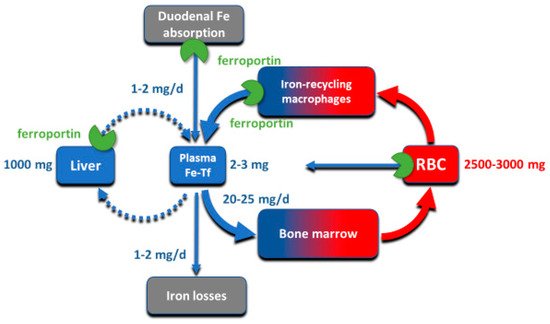
Although all cells in a multicellular organism contain iron, systemic iron homeostasis is primarily affected by the following compartments: the erythron (red blood cells and their precursors in erythropoietic organs), two types of stores (hepatocytes of the liver and macrophages of the spleen and the liver), blood plasma which moves iron between tissues and organs, and absorptive enterocytes in the duodenum through which iron enters the body, ordinarily to replace small losses from the body caused by shedding of iron-containing cells ().
Figure 1. The key iron flows and compartments.
The largest of these iron compartments is the erythron (erythrocytes and their precursors), which contains approximately 2–3 g of iron, representing about 2/3–3/4 of the total body iron in adult humans. Erythroid iron is almost entirely contained within hemoglobin, at the concentration of 1 g of iron per liter of packed erythrocytes. The second largest compartment are the hepatocyte stores, where up to 1 g of iron is contained in cytoplasmic ferritin of hepatocytes. The stores are highly variable and can be nearly depleted in many women of reproductive age because of menstrual blood loss combined with low dietary intake. The recycling macrophages in the spleen, liver and marrow function as a rapid turnover compartment
[1]. The spleen, which represents a substantial portion of the macrophage storage compartment, normally contains only about 0.05 g of iron but has the capacity for much larger amounts
[2]. Only about 0.3 g of iron are contained in the other tissues, in myoglobin of muscles and in iron-containing enzymes.
Iron is transported around the body on blood plasma carrier protein transferrin, whose two iron-binding sites are normally 20–45% occupied. Each cell in the body has transferrin receptors (TfR1) that are endocytosed into acidified endosomes where iron dissociates from the TfR1-transferrin complex and is transported across the endosomal membrane into the cytoplasm. The transferrin compartment in blood plasma holds only 2–3 mg of iron but delivers to target tissues 20–25 mg/day so its iron content turns over every few hours. Despite changes in dietary iron content and tissue demand for iron, the concentration of iron in the blood plasma in healthy humans is remarkably stable, as is the total body iron content. Moreover, duodenal iron absorption increases several-fold after blood loss or exposure to hypoxia, or in response to iron deficiency. These observations provided early evidence that the absorption and tissue distribution of iron must be subject to endocrine regulation
[3].
3. Molecular Basis of Systemic Iron Homeostasis
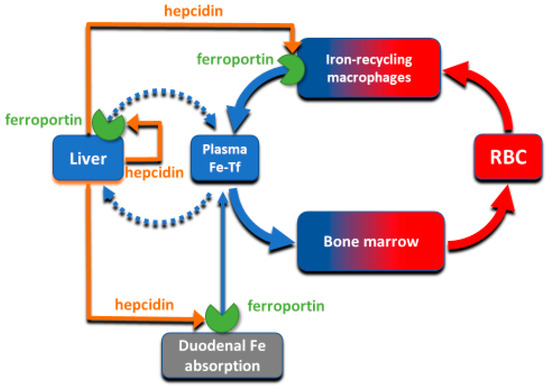
The absorption and tissue distribution of iron is principally controlled by the interaction of the hepatic hormone hepcidin with ferroportin. Ferroportin is expressed in iron-storing and iron-transporting tissues
[4] and functions both as the hepcidin receptor and the sole known cellular exporter of elemental iron in multicellular organisms (). The 25 amino-acid hepcidin peptide (MW 2.8 kD) is synthesized by hepatocytes and secreted into blood plasma, with concentrations in healthy humans ranging from approximately 2–20 nM
[5][6][5,6], around hundred-fold higher than the concentration of the similarly-sized peptide hormones insulin, glucagon or parathyroid hormone.
Figure 2. The interaction of hepcidin with ferroportin controls iron flows into plasma.
4. Ferroportin Structure
Ferroportin, a member of the solute carrier family, is systematically named SLC40A1. The human protein has 571 amino acids for a molecular weight of around 65–70 kD
[7][8][9][7,8,9]. The variation in molecular mass is likely caused by tissue-specific glycosylation but the functional consequences of the glycosylation differences between the forms purified from the duodenal enterocytes, hepatocytes and macrophages are not yet understood
[10]. Structurally, ferroportin consists of two 6-transmembrane-helix bundles (N-lobe and C-lobe) joined by a cytoplasmic loop, with both C- and N-termini located in the cytoplasm (). The two helix bundles enclose a cavity through which iron is thought to exit the cell. The shape and similarity to more completely characterized family members suggest that ferroportin exports cellular iron by an alternating access mechanism, wherein ferroportin alternates between an open-inward conformation which binds intracellular iron, and an open-outward conformation which releases the iron to the extracellular space (). The remarkable feat of structural characterization of a ferroportin from the bacterium
Bdellovibrio bacteriovorus in both open-outward and open-inward conformations
[11] represents strong support for such a model. The exported species is almost certainly ferrous iron
[12] but the energetics of iron export is not yet well characterized. Because iron is exported against the force exerted by the transmembrane electric field, and there is no evidence of direct coupling of the transport to ATP hydrolysis, the export of iron is likely facilitated by the coupled “downhill” transport of another ion or small molecule
[13].
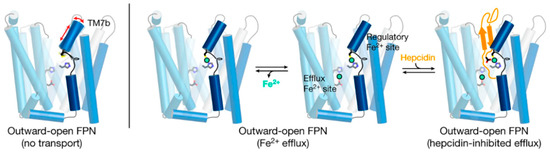
Figure 3. A current model of hepcidin (orange) interaction with ferroportin (blue) in which binding is dependent on iron (green). The framework of helices that make up ferroportin are depicted as cylinders connected by extracellular and intracellular disordered loops. The side chains that make up the binding sites for iron are shown. Modified from
[14].
Structural analyses of ferroportin indicate that there are two divalent metal-binding sites, one in each lobe, facing the internal cavity of ferroportin ()
[11][13][14][11,13,14]. How these sites mediate the export of iron is not yet clear. The site in the N-lobe can bind calcium which may have a modulatory role on iron transport
[13]. Although calcium itself is not transported, it is required for human ferroportin transport activity, and is thought to directly bind to ferroportin and facilitate a conformational change critical to the transport cycle. Another area of interest is how cytoplasmic iron destined for export reaches ferroportin. Current evidence indicates that cytoplasmic iron is predominantly present as a complex of ferrous iron with reduced glutathione (summarized in
[15]), and that the complex of ferrous iron and reduced glutathione may be delivered to ferroportin via the cytoplasmic iron chaperone PCBP2
[16].
5. Ferroportin Interactions with Hepcidin
The flow of iron out of the cells is controlled by hepcidin through two known mechanisms: occlusion of the open-outward conformation of ferroportin by hepcidin
[17], and hepcidin-induced endocytosis and degradation of ferroportin
[4]. The occlusion mechanism would be effective at hepcidin concentrations where most ferroportin molecules remain occluded by hepcidin most of the time, and would be rapidly reversible when hepcidin concentrations are decreased. The endocytosis mechanism could be initiated by even transient binding of hepcidin to ferroportin, and would cause the permanent removal of ferroportin from the cell surface that would require resynthesis of ferroportin for the recovery of iron transport. The second mechanism would therefore be expected to occur at lower concentrations of hepcidin, and to have a prolonged effect, even if hepcidin concentrations subsequently decrease. Although hepcidin peptide injected into mice is cleared from blood circulation within minutes, its plasma iron lowering effect lasts for 24–48 h, supporting the importance of the endocytic mechanism for ferroportin physiology
[18].
Hepcidin regulation of ferroportin by the endocytic mechanism resembles the generic ligand-induced receptor endocytosis. It appears to require hepcidin-induced conformational change in ferroportin that triggers the ubiquitination of the lysine-rich cytoplasmic segment connecting the two 6-helix domains of ferroportin
[19][20][19,20]. Ubiquitinated ferroportin is then targeted to lysosomes and proteasomes for degradation. A recent study
[21] provide convincing evidence that
Rnf217 is an important E3 ubiquitin ligase that triggers the degradation of ferroportin in response to hepcidin binding.
Recent advances in structural understanding of the hepcidin-ferroportin interaction confirmed the simple models generated by targeted mutagenesis of hepcidin and ferroportin but also provided surprising new details
[14]. In a nanodisc membrane model, hepcidin is seen to bind to the C-lobe of ferroportin (), with the highly variable hepcidin loop largely extracellular, and the relatively conserved C- and N-termini of hepcidin deeply buried in the central cavity. The model also revealed an important unexpected feature, the indication that the suspected hepcidin-binding site in the C-lobe, centered on the critically important C325 thiol cysteine, utilizes an iron atom to coordinate hepcidin binding
[14]. It is not yet clear how this contributes to hepcidin physiology but it can be anticipated that the mechanism could provide selectivity for hepcidin to bind to ferroportin molecules actively transporting iron as opposed to those that may be in a resting state. One indication that this mechanism may have a physiological effect is the recently reported inhibition of hepcidin-induced ferroportin degradation by the intracellular iron chelator deferiprone but not by the extracellularly acting chelator deferoxamine
[22]. How the new structural model accommodates other hepcidin agonists ranging from minihepcidin peptides
[23] to small molecules
[24] remains to be determined.