Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sandra Louzada and Version 2 by Vivi Li.
Repetitive DNA is a major organizational component of eukaryotic genomes, being intrinsically related with their architecture and evolution. Tandemly repeated satellite DNAs (satDNAs) can be found clustered in specific heterochromatin-rich chromosomal regions, building vital structures like functional centromeres and also dispersed within euchromatin. Interestingly, despite their association to critical chromosomal structures, satDNAs are widely variable among species due to their high turnover rates. This dynamic behavior has been associated with genome plasticity and chromosome rearrangements, leading to the reshaping of genomes.
- satellite DNA
- genome architecture
- chromosome restructuring
- Robertsonian translocations
- satellite DNA transcription
1. Introduction
The linear organization of DNA sequences in the genome and how these sequences are packed into chromosomes define their architecture and influence its evolution. Repetitive DNA represents a major organizational component of eukaryotic genomes and includes sequences dispersed throughout the genome like transposable elements (TEs) and tandemly repeated sequences, such as satellite DNA (satDNA) [1][2][1,2]. Together with TEs, satDNAs contribute significantly to the differences in genome size between species, accounting for more than 50% of some species total DNA [3]. SatDNAs can be found in varied locations in the chromosomes, such as pericentromeric, subtelomeric and interstitial regions, forming blocks of constitutive heterochromatin (CH) [2][3][4][5][6][7][2,3,4,5,6,7] that are part of vital structures like centromeres and telomeres [2]. However, satDNA location is not restricted to CH with some satDNAs being found also dispersed throughout euchromatic regions in different species [5][8][5,8]. Multiple lines of evidence show that satDNAs have key roles in centromere function, heterochromatin formation and maintenance and chromosome pairing [9][10][11][12][9,10,11,12]. Interestingly, despite their association to critical chromosomal structures, satDNA families can display an astounding sequence variation even among closely related species. This results from their highly dynamic behavior, leading to rapid changes in sequence composition and array size within short evolutionary periods, which can lead to speciation (reviewed in [13]). Moreover, these sequences have been consistently correlated with fragile sites and evolutionary breakpoint regions in diverse species [14][15][16][17][18][19][14,15,16,17,18,19] and are intrinsically involved in frequent chromosomal rearrangements like Robertsonian translocations [20][21][20,21]. SatDNA dynamics has been shown to promote genome plasticity and to have an active involvement in the modulation of genomic architecture by promoting rearrangements.
Nevertheless, some satDNAs seem to have been preserved or “frozen” across different taxa during long evolutionary periods [22][23][24][22,23,24] with some of them being transcribed into satellite non-coding RNAs (satncRNAs). Indeed, transcripts of satDNAs have been reported in different species, highlighting a possible role for satncRNAs in the regulation of gene expression, cancer outcomes and aging [25][26][27][25,26,27]. This suggests that functional constraints may be causing the preservation of these sequences over the time [24][28][24,28]. Accordingly, some species centromeric satDNAs have been found to share a 17 bp motif known as the centromere protein B (CENP-B) box, representing the binding site for centromere protein B (CENP-B) [29][30][29,30]. It has been demonstrated that the CENP-B box is required for de novo centromere chromatin assembly and CENP-B protein is involved in centromere functions [31]. In this case, the conservation of a sequence motif across diverse mammalian species satDNAs [32] seems to be related to a specific function.
2. SatDNA Features and Organization in the Genome and Chromosomes: Emerging Technologies and Changing Concepts
The concept of satDNA suffered considerable changes through time. Early experiments historically coined the term “satellite DNA” referring to tandemly arranged sequences that formed satellite bands separate from the rest of the genomic DNA during density gradient centrifugation [33][36]. Given that no function was initially attributed to these sequences, they were considered as genomic “junk”, representing parasites proliferating independently in the genomes [34][37]. Today, satDNAs are viewed as important genomic functional components. In order to understand participation of these sequences in genome architecture and evolution, we need to briefly address their organizational features, localization and mode of evolution. SatDNA is typically organized as long arrays of head-to-tail linked repeats and usually present in the genomes in several million copies [1]. The length of the repeating unit (monomer) can range from a few base pairs up to more than 1 kb, forming arrays that may reach 100 Mb in length (reviewed by [35][38]), and that can form higher-order repeat (HOR) units (e.g., [36][37][38][39,40,41]). Human chromosome centromeres are populated by α satDNA (αSAT) organized in HORs that are structurally distinct and confer chromosome specificity [36][39][39,42]. Complex HORs have been found in non-human mammals such as insects, mouse, swine, bovids, horse, dog and elephant (reviewed in [40][43]), and more recently in Callitrichini monkeys [41][44] and Teleostei fish [42][45]. SatDNA arrays are mainly found clustered in heterochromatin, although studies also report the presence of short satDNA arrays dispersed along euchromatic regions [2][3][4][5][6][7][2,3,4,5,6,7]. These sequences can be found in varied locations in the chromosomes, such as pericentromeric, subtelomeric and interstitial regions [2][43][44][45][2,46,47,48], as well as being part of vital structures like centromeres and telomeres [2]. Usually more than one family of satDNAs can be found in the same genome, thus forming a library, which can be shared among closely related species. The satDNAs within the library may differ in monomer sequence, size, abundance, distribution and location (reviewed in [12]). Expansions and contractions of satDNA arrays can dramatically change the landscape of repetitive sequences, leading to significant differences of satDNA copy number among related species [46][47][49,50]. That is the case of the Drosophila genus, which contains very dissimilar satDNAs, varying from 0.5% in some species genomes to as high as 50% in others [48][49][51,52]. Such striking differences in satDNA abundance in Drosophila sp. were proposed to result predominantly from lineage-specific gains accumulated over the past 40 MY of evolution [50][53], ultimately causing species reproductive barriers [51][52][54,55]. The mechanisms proposed to be responsible for the amplification/deletion of repetitive DNA, consequently leading to their rapid evolutionary turnover, are unequal crossing over, replication slippage and rolling circle amplification [53][56]. SatDNA sequence divergence among species is quite variable, as some repeats are species-specific, while others are widely conserved, being shared across distantly related species [22][24][54][22,24,57]. SatDNAs have a unique mode of evolution, known as concerted evolution, a two-level process in which mutations are homogenized throughout monomers of a repetitive family and concomitantly fixed within a group of reproductively linked organisms [55][56][58,59]. The study and characterization of satDNA has lagged behind when compared with other genomic sequences. Throughout time, different methodological approaches have generated insights into the structure, organization, function and evolution of these sequence elements, although this characterization has been significantly hampered by their highly repetitive nature. The advent of high-throughput sequencing technologies and associated bioinformatics tools opened the door to whole genome sequencing projects, and as the technology became more robust and cheaper, the number of sequenced species increased exponentially. In 2018, the Earth BioGenome project was launched, aiming to increase the number of sequenced eukaryotic genomes from 2534 species (of which only 25 comply with the standard for contig and scaffold N50 established by the Genome 10K organization) to characterize the genomes of the 1.5 million known species within a 10 year time frame [57][60]. Of note, satDNA, as well as other repetitive sequences, have been systematically omitted from the genome projects, due to difficulties in sequence alignment and assembly, given that the read length of current sequencing technologies is unable to span the longer repeats and tandem arrays [58][59][61,62]. Nevertheless, high-throughput sequencing contributed significantly to increase our knowledge regarding satDNA sequences [60][63]. Next generation sequencing (NGS; e.g., Illumina), allied to newly developed bioinformatics tools capable of identifying satDNA sequences in unassembled data (e.g., RepeatExplorer) [61][62][63][64,65,66], helped uncover the extent of satDNAs present in the genome of different species, revealing unpredicted levels of satDNA diversity (e.g., [64][65][66][67][68][69][34,67,68,69,70,71]). For instances, 62 satDNA families were identified in the genome of the migratory locust, leading to the coining of the term ‘satellitome’ to refer to the whole collection of satDNA families found in a single genome [64][34], a part of the ‘repeatome’, a term proposed previously [70][33] to refer to the collection of all repetitive sequences in a genome (TEs, satDNAs, etc.). This number has been surpassed by a recent study where 164 satDNA families have been identified in Teleostei fish, being this the biggest satellitome characterized for a given species so far [68][70]. The availability of a methodology capable of assessing satDNA array abundance and diversity led to an explosion of comparative studies across a wide range of clades, including mammals, insects and plants (e.g., [41][42][67][69][71][72][44,45,69,71,72,73]) providing insights into these sequences. The development of sequencing technologies that generate long-range data has allowed the community to overcome some of the limitations imposed by NGS and is fueling the study of repeats. Single-molecule real-time sequencing and nanopore sequencing technologies (commercialized by PacBio and Oxford Nanopore Technologies (ONT), respectively) can generate longer reads capable of spanning repetitive regions, thus enabling their assembly into contigs (reviewed in [59][62]). For instances, ONT nanopore sequencers have been shown to generate unprecedented ultra-long reads that can reach mega-base lengths, leading to significant improvements in the human genome assembly [73][74][75][76][74,75,76,77], with some of the repetitive-containing gaps being closed [77][78][78,79]. By using long-read methods we are gaining access to important repeated-rich structures, like centromeres, revealing further insights into their sequence content and structure [79][80]. For instances, Drosophila centromeric satDNAs were recently shown to be intermingled with TEs [80][81]. Other recent studies report the improvement of human Y chromosome centromere assembly [77][78] and the reconstruction of a 2.8 megabase centromeric satDNA array, with the potential to achieve for the first-time telomere-to-telomere sequencing of the X chromosome [78][79]. Several studies demonstrate that the combination of different high-throughput sequencing methods (e.g., Illumina, ONT and PacBio) with other techniques, such as optical mapping, cytogenetics and molecular techniques, is beneficial and sometimes essential to determine satDNA features. The use of PacBio long-read sequencing together with optical mapping proved to be helpful in the assembly of satDNA arrays with large monomers and provided insights regarding recombination rates in the Eurasian crow [81][82]. Positional data derived from fluorescent in situ hybridization (FISH) remains vital to determine the physical location of satDNAs, since such information cannot be achieved for genomes that have not yet been properly assembled (e.g., [64][41][69][80][82][34,44,71,81,83]), and sequences mapping by FISH on extended DNA fibers can provide significant assistance to the process of genome assembly, aiding in contig ordering (e.g., [83][84][84,85]). Improved techniques based on FISH, helped shedding light into repetitive-rich chromosome regions with centromeric function (e.g., [85][86]). Other methods have also shown to provide a valid and expedite analysis of repetitive sequences profile, such as PCR-based approaches, that have been used to determine satDNA copy number differences between healthy and cancer cells/tissues [86][87]. In particular, the use of droplet digital PCR (ddPCR) combined with other methodologies has contributed to the validation and quantification of rare retrotransposon insertion events in different tissues including tumors [87][88] and the detection and accurate quantification of human SATII ncRNA in cancer patients [88][89]. The integration of genomic, cytogenetic and cell biology data helps to establish a connection between sequence information, its localization in the chromosomes and their interaction with other components of the genome, defining the field of chromosomics [89][90]. We believe that this approach is essential to fully understand the organization of repetitive sequences. Other aspects of satDNA biology are also becoming accessible through the use of recent methodologies, such as the characterization of their expression and chromatin state, namely by using RNA sequencing (RNA-seq) and chromatin immunoprecipitation approaches followed by DNA sequencing (CHIP-seq) [90][91][91,92]. In particular, for CHIP-seq experiments several studies report the use of a specific antibody against DNA binding centromere-specific histone H3 (CENH3), which is an ortholog for human CENP-A. This methodology has proven to be useful for clarifying the satDNA content in the centromere, improving some organisms reference sequence and uncovering satDNA variability (e.g., [92][93][93,94]). The data generated is now being used to determine satDNA sequences organization in the genome [94][95], explore predicted evolutionary patterns and hypothesis (e.g., [95][66][96][97][35,68,96,97]), as well as to shed light into the function of these sequences [80][98][81,98]. We are now closer than ever to fully access the sequence information hidden within repetitive-rich chromosome structures like centromeres and telomeres. However, we still need to further develop and adapt currently available approaches to achieve a combination of genomic, cytogenetic and molecular techniques to optimally address these regions, which we propose could be referred to as centrOMICs and telOMICs (Figure 1). SatDNAs represent one of the most intriguing and also interesting components of the genome and their full characterization will help us to better understand genome organization, architecture and evolution.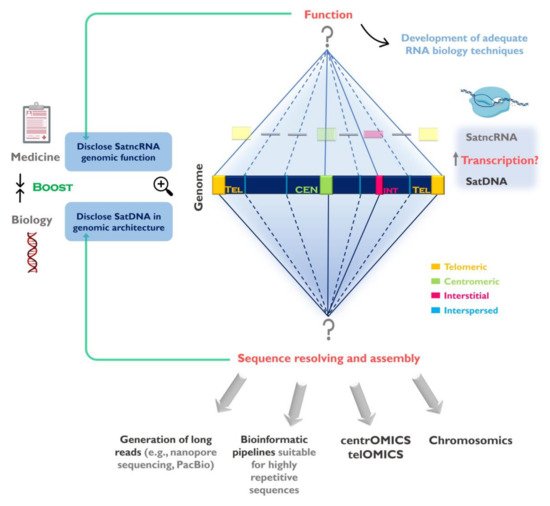
Figure 1. Challenges in the study of satellite DNA (satDNA) sequences and the importance to fully understand the repetitive genomic fraction. SatDNAs can be found clustered at the centromeres, telomeres and forming interstitial heterochromatin (CH) blocks, as well as scattered (interspersed) throughout the chromosomes. The full characterization of satDNAs needs to be addressed in two levels: 1-Disclose satDNAs linear sequence and improve their representation in genome assemblies. Despite currently used sequencing strategies (e.g., next generation sequencing (NGS)) contributed for satDNA studies, the full characterization of these sequences will only be achieved by using sequencing technologies capable of long reads, bioinformatics pipelines suitable for highly repetitive sequences, together with other techniques (e.g., FISH, optical mapping). These strategies need to be directed to specific chromosome structures such as centromeres (centrOMICs) and telomeres (telOMICs), which harbor large amounts of satDNA. Important also is the integration of genomic data with sequence localization in the chromosomes, and their interaction with other components of the genome (chromosomics); 2- Clarify satDNAs function(s) in the genome by studying the satellite non-coding RNAs (satncRNA) and their interaction with other components and structures in the genome. In this field there is the need to develop adequate biology techniques to address repetitive sequences transcription study. The disclosure of satDNA sequences will help to better understand its genomic architecture ant its role in genome restructuring in evolution and disease.