Lipids and lipoproteins constitute indispensable components for living not only for humans. In the case of hepatitis C virus (HCV), the option of using the products of our lipid metabolism is “to be, or not to be”. On the other hand, HCV infection, which is the main cause of chronic hepatitis, cirrhosis and hepatocellular carcinoma, exerts a profound influence on lipid and lipoprotein metabolism of the host.
- HCV
- lipoproteins
- lipoprotein receptors
- dislipidemia
- apolipoproteins
1. Introduction
Hepatitis C virus (HCV), belonging to the family of Flaviviridae, genus
Hepacivirus
, is a small-enveloped virus with a single (+)-stranded RNA genome of approximately 9.5 kb (
Figure 1). It is estimated that over 71 million people worldwide are infected with HCV, accounting for 1% global population [6]. A majority of infected people do not realize that they have been infected, and the silently developing infection remains a major causative agent of chronic liver diseases. In turn, chronic hepatitis C (CHC) has been shown to be responsible for up to 25% of hepatocellular carcinoma cases worldwide [7]. Persistence and pathogenesis of such viral infection are caused by the ability of HCV to deregulate important host processes, mostly innate immunity and lipid metabolism. The association of HCV with lipid and lipoprotein metabolism has been observed clinically for many years. In particular, chronic hepatitis is considered to be the reason of liver steatosis and dyslipidemia [8,9].
). It is estimated that over 71 million people worldwide are infected with HCV, accounting for 1% global population [1]. A majority of infected people do not realize that they have been infected, and the silently developing infection remains a major causative agent of chronic liver diseases. In turn, chronic hepatitis C (CHC) has been shown to be responsible for up to 25% of hepatocellular carcinoma cases worldwide [2]. Persistence and pathogenesis of such viral infection are caused by the ability of HCV to deregulate important host processes, mostly innate immunity and lipid metabolism. The association of HCV with lipid and lipoprotein metabolism has been observed clinically for many years. In particular, chronic hepatitis is considered to be the reason of liver steatosis and dyslipidemia [3][4].
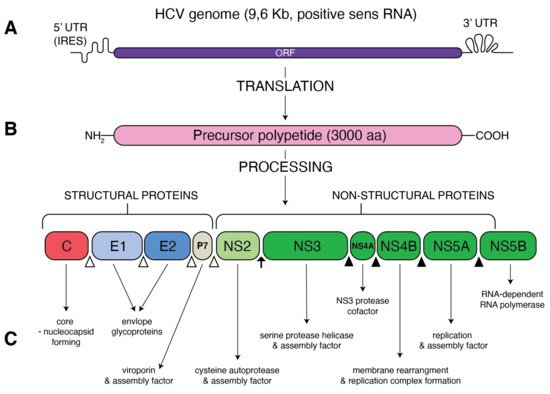
Figure 1.
Schematic presentation of HCV genome and proteins. (
A
) A single open reading frame (ORF) of HCV genome is flanked by 5′ untranslated region (5′ UTR) that contains an internal ribosome entry site (IRES) and 3′ UTR. (
B
) IRES-mediated translation forms a precursor protein of 3000 aa, which is then processed into 10 different proteins. Structural proteins (core, E1, E2 and p7) are cleaved from precursor thanks to the cellular signal peptidase (empty triangles) The NS2–NS3 protease auto-cleaves itself (black arrow). The NS3 protease, with NS4A as a cofactor, cleaves the remaining non-structural proteins: NS3, NS4A, NS4B, NS5A and NS5B (black triangles). (
C
) Function of the resultant structural and non-structural proteins.
2. Lipid Phenotype of Chronic HCV Patients
Immediately after the diagnostic tests for HCV became available, it was stated that hepatic steatosis is one of the characteristic histological features in the liver of HCV-infected patients. HCV-infected people have significantly higher prevalence of steatosis than in hepatitis B virus (HBV)-infected patients, and it is particularly strong in Genotype 3 infection, demonstrating a correlation between genetic variation of HCV and the ability to accumulate lipids [14,15]. The pathogenesis of HCV-induced steatosis is determined by viral factors as well as host factors.
Immediately after the diagnostic tests for HCV became available, it was stated that hepatic steatosis is one of the characteristic histological features in the liver of HCV-infected patients. HCV-infected people have significantly higher prevalence of steatosis than in hepatitis B virus (HBV)-infected patients, and it is particularly strong in Genotype 3 infection, demonstrating a correlation between genetic variation of HCV and the ability to accumulate lipids [5][6]. The pathogenesis of HCV-induced steatosis is determined by viral factors as well as host factors.
3. Resemblance between HCV Particles and Lipoproteins
In general terms, the HCV particle should consist of a nucleocapsid containing the single-stranded RNA genome connected to the viral core protein and an envelope membrane containing the surface viral glycoproteins E1 and E2 [35]. However, viral particles purified from sera of infected patients revealed that the structure of HCV is much more complex and HCV particles are extremely heterogeneous [36].
In general terms, the HCV particle should consist of a nucleocapsid containing the single-stranded RNA genome connected to the viral core protein and an envelope membrane containing the surface viral glycoproteins E1 and E2 [7]. However, viral particles purified from sera of infected patients revealed that the structure of HCV is much more complex and HCV particles are extremely heterogeneous [8].
HCV particles in the serum are presented as a mix of complete low-density infectious, so-called lipo-viral particles (LVP), nonenveloped nucleocapsids and a great amount of empty lipoviral particles (eLVP) that are also described as nucleocapsid-free sub-viral particles (
Figure 2). LVP is known as a hybrid particle that consists of viral components and cell-derived triglyceride-rich lipoproteins, that include some apolipoproteins (ApoE, ApoB, ApoCI, ApoCII and ApoCIII). In comparison to lipoproteins of the same density, LVP have 30 times more TG per particle. This is probably because of a heavier non-lipid load consisting of HCV RNA and the core protein [44]. An analysis of sucrose gradients in the serum of infected people showed that HCV RNA is distributed over a variety of densities from 1.20 to 1.03 g/cm
). LVP is known as a hybrid particle that consists of viral components and cell-derived triglyceride-rich lipoproteins, that include some apolipoproteins (ApoE, ApoB, ApoCI, ApoCII and ApoCIII). In comparison to lipoproteins of the same density, LVP have 30 times more TG per particle. This is probably because of a heavier non-lipid load consisting of HCV RNA and the core protein [9]. An analysis of sucrose gradients in the serum of infected people showed that HCV RNA is distributed over a variety of densities from 1.20 to 1.03 g/cm
3 [34]. Virions with a very low buoyant density (range 1.10–1.14 g/mL) have shown the highest infectivity [45]. The contribution of LVP to total HCV viral load is highly dynamic and affected by lipoprotein metabolism [44]. The presence of nonenveloped HCV nucleocapsids in the serum of HCV-infected patients probably contributes to persistent infection and HCV escape from immune surveillance [46]. Apart from infectious LVP, envelope E1 and E2 proteins were found on the surface of lipoprotein particles devoid of infectious nucleocapsids, also known as empty LVP. Their presence may contribute to the pathology of hepatitis C [44]. The presence of apolipoproteins on the surface of LVP and empty LVP can mask viral epitops and allows HCV to avoid anti-HCV neutralizing antibodies [47].
[10]. Virions with a very low buoyant density (range 1.10–1.14 g/mL) have shown the highest infectivity [11]. The contribution of LVP to total HCV viral load is highly dynamic and affected by lipoprotein metabolism [9]. The presence of nonenveloped HCV nucleocapsids in the serum of HCV-infected patients probably contributes to persistent infection and HCV escape from immune surveillance [12]. Apart from infectious LVP, envelope E1 and E2 proteins were found on the surface of lipoprotein particles devoid of infectious nucleocapsids, also known as empty LVP. Their presence may contribute to the pathology of hepatitis C [9]. The presence of apolipoproteins on the surface of LVP and empty LVP can mask viral epitops and allows HCV to avoid anti-HCV neutralizing antibodies [13].
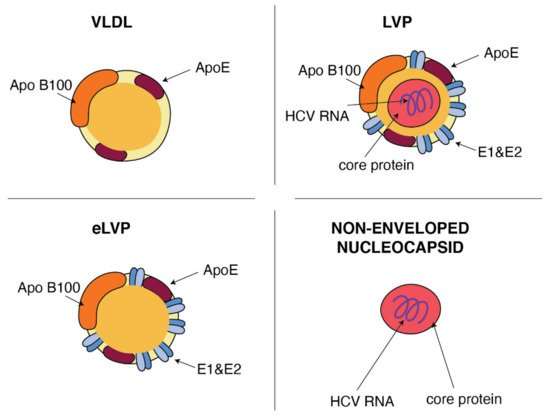
Figure 2.
Similarities between VLDL and HCV particles. ApoB and ApoE apolipoproteins classically associated with host VLDL particles are very useful components of HCV particles: lipo-viral particles (LVP) and empty lipo-viral particles (eLVP). An additional component that appears in the lipid envelope of these two HCV particles is a heterodimer composed of viral E1 and E2 envelope glycoproteins. Capsid, formed by the viral core protein, containing the positive HCV RNA strand may be surrounded by the lipid envelop with embedded E1 and E2 glycoproteins, such as in the case of LVP, or may form a non-enveloped viral particle also presented in the blood of HCV-infected patients.
4. Lipid-Dependent HCV Entry
HCV entry into human hepatocytes is a multi-step process (
Figure 3) in which many host factors are involved including glycoaminoglycans (GAGs), low density lipoprotein receptors (LDLr), scavenger receptors class B type I (SR-BI), tetraspanin CD81, the tight junction proteins, claudin-1 (CLDN1), occludin (OCLN), receptor tyrosine kinases (RTKs), the Niemann–Pick C1-like 1 (NPC1L1) and epidermal growth factor receptor (EGFR) [55,56].
) in which many host factors are involved including glycoaminoglycans (GAGs), low density lipoprotein receptors (LDLr), scavenger receptors class B type I (SR-BI), tetraspanin CD81, the tight junction proteins, claudin-1 (CLDN1), occludin (OCLN), receptor tyrosine kinases (RTKs), the Niemann–Pick C1-like 1 (NPC1L1) and epidermal growth factor receptor (EGFR) [14][15].
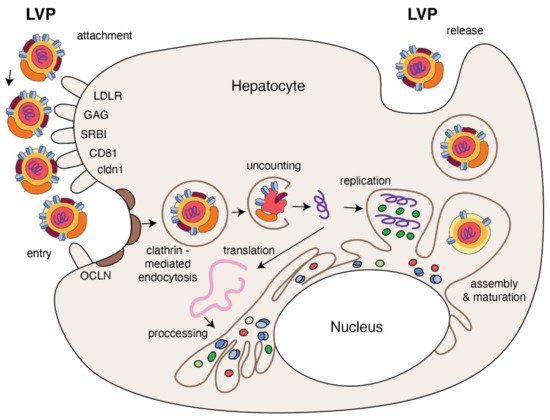
Figure 3.
The replication cycle of HCV. Lipo-viral particle binds to the host cellular receptors such as LDLR, SRB1, CD81, claudin1 and others (please refer to the main text). Following attachment, the virus enters the target cell via clathrin-mediated endocytosis. Upon entry, HCV RNA is released into the cytoplasm and is directly translated via an IRES at the ER. A polyprotein precursor is then processed by both viral and host proteases. Non-structural viral proteins and host factors form a membranous web and replication complexes that are used for synthesis of multiple HCV RNA copies. Viral progenies are packed, forming nucleocapsid that is enriched by viral envelope glycoprotein. The access to the lipoprotein pathway provides conditions for incorporation of neutral lipids and apolipoproteins and formation of the mature lipo-viral particle released from hepatocytes by exocytosis.
5. Host Factors Contribution to the Efficient HCV Replication
Replication is the next complex process of the viral life cycle that is facilitated by interaction of HCV with various lipid-related factors. CV RNA replication requires geranylgeranylated host proteins, as was found for prenylated host FBL2 protein that binds to NS5A [84]. It may explain how NS5A protein colocalizes in membrane, where HCV RNA replication occurs. Newly synthetized HCV proteins, together with host factors, induce the formation of special platform, the so-called membranous web (MW), composed of vesicles as well as cytosolic lipid droplets (LD) [85]. HCV, similar to other positive-sense RNA viruses, remodels intracellular membranes in host cells to form an effective replication site. For this reason, HCV nonstructural proteins function to modify the endoplasmic reticulum and cytosolic lipid droplets to form double membrane vesicles (DMVs) containing HCV replication complexes [85] where viral RNA is protected from host RNAses and sensors of the innate immunity that would respond to double-stranded RNA [85,86]. HCV replication complexes contain HCV RNA, the non-structural viral proteins necessary for replication and the newly synthesized viral RNA [87,88]. Massive rearrangement of host lipid structure is indispensable to form efficient HCV replication complexes. The study on lipin 1 [89], an enzyme that forms diacylglycerol in the cytoplasm and regulates the expression of genes involved in lipid metabolism, showed the involvement of this enzyme in generation of membranous web. It suggests the capacity of lipin1 to modify lipids and contribute to the formation of MW. Resulting membranous is composed mostly from double-membrane vesicles. These cholesterol-enriched membranes are formed by lipid transfer proteins such as Nieman–Pick type C1 protein that requited cholesterol to the membranous replication organelle. It was observed that NPC1 inhibition alters cholesterol transport and reduces HCV replication [90].
Replication is the next complex process of the viral life cycle that is facilitated by interaction of HCV with various lipid-related factors. CV RNA replication requires geranylgeranylated host proteins, as was found for prenylated host FBL2 protein that binds to NS5A [16]. It may explain how NS5A protein colocalizes in membrane, where HCV RNA replication occurs. Newly synthetized HCV proteins, together with host factors, induce the formation of special platform, the so-called membranous web (MW), composed of vesicles as well as cytosolic lipid droplets (LD) [17]. HCV, similar to other positive-sense RNA viruses, remodels intracellular membranes in host cells to form an effective replication site. For this reason, HCV nonstructural proteins function to modify the endoplasmic reticulum and cytosolic lipid droplets to form double membrane vesicles (DMVs) containing HCV replication complexes [17] where viral RNA is protected from host RNAses and sensors of the innate immunity that would respond to double-stranded RNA [17][18]. HCV replication complexes contain HCV RNA, the non-structural viral proteins necessary for replication and the newly synthesized viral RNA [19][20]. Massive rearrangement of host lipid structure is indispensable to form efficient HCV replication complexes. The study on lipin 1 [21], an enzyme that forms diacylglycerol in the cytoplasm and regulates the expression of genes involved in lipid metabolism, showed the involvement of this enzyme in generation of membranous web. It suggests the capacity of lipin1 to modify lipids and contribute to the formation of MW. Resulting membranous is composed mostly from double-membrane vesicles. These cholesterol-enriched membranes are formed by lipid transfer proteins such as Nieman–Pick type C1 protein that requited cholesterol to the membranous replication organelle. It was observed that NPC1 inhibition alters cholesterol transport and reduces HCV replication [22].
6. HCV Hijacks the VLDL Secretory Pathway for Progeny Formation
The special membranous matrix formation is not only necessary for HCV RNA replication, rearrangement of lipid membranes and lipid droplets in host cell is also a prerequisite for a successful viral assembly. Probably, to avoid competition for using HCV RNA, HCV replication is separate in time and space from the formation of nucleocapsid [101].
The special membranous matrix formation is not only necessary for HCV RNA replication, rearrangement of lipid membranes and lipid droplets in host cell is also a prerequisite for a successful viral assembly. Probably, to avoid competition for using HCV RNA, HCV replication is separate in time and space from the formation of nucleocapsid [23].
References
- Roudot-Thoraval, F. Epidemiology of hepatitis C virus infection. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101596.
- Sagnelli, E.; Macera, M.; Russo, A.; Coppola, N.; Sagnelli, C. Epidemiological and etiological variations in hepatocellular carcinoma. Infection 2020, 48, 7–17.
- Bamber, M.; Murray, A.K.; Weller, I.V.; Morelli, A.; Scheuer, P.J.; Thomas, H.C.; Sherlock, S. Clinical and histological features of a group of patients with sporadic non-A, non-B hepatitis. J. Clin. Pathol. 1981, 34, 1175–1180.
- Fabris, C.; Federico, E.; Soardo, G.; Falleti, E.; Pirisi, M. Blood lipids of patients with chronic hepatitis: Differences related to viral etiology. Clin. Chim. Acta 1997, 261, 159–165.
- Sharma, P.; Balan, V.; Hernandez, J.; Rosati, M.; Williams, J.; Rodriguez-Luna, H.; Schwartz, J.; Harrison, E.; Anderson, M.; Byrne, T.; et al. Hepatic steatosis in hepatitis C virus genotype 3 infection: Does it correlate with body mass index, fibrosis, and HCV risk factors? Dig. Dis. Sci. 2004, 49, 25–29.
- Thomopoulos, K.C.; Arvaniti, V.; Tsamantas, A.C.; Dimitropoulou, D.; Gogos, C.A.; Siagris, D.; Theocharis, G.J.; Labropoulou-Karatza, C. Prevalence of liver steatosis in patients with chronic hepatitis B: A study of associated factors and of relationship with fibrosis. Eur. J. Gastroenterol. Hepatol. 2006, 18, 233–237.
- Lavillette, D.; Pécheur, E.I.; Donot, P.; Fresquet, J.; Molle, J.; Corbau, R.; Dreux, M.; Penin, F.; Cosset, F.L. Characterization of fusion determinants points to the involvement of three discrete regions of both E1 and E2 glycoproteins in the membrane fusion process of hepatitis C virus. J. Virol. 2007, 81, 8752–8765.
- Pumeechockchai, W.; Bevitt, D.; Agarwal, K.; Petropoulou, T.; Langer, B.C.; Belohradsky, B.; Bassendine, M.F.; Toms, G.L. Hepatitis C virus particles of different density in the blood of chronically infected immunocompetent and immunodeficient patients: Implications for virus clearance by antibody. J. Med. Virol. 2002, 68, 335–342.
- Scholtes, C.; Ramière, C.; Rainteau, D.; Perrin-Cocon, L.; Wolf, C.; Humbert, L.; Carreras, M.; Guironnet-Paquet, A.; Zoulim, F.; Bartenschlager, R.; et al. High plasma level of nucleocapsid-free envelope glycoprotein-positive lipoproteins in hepatitis C patients. Hepatology 2012, 56, 39–48.
- Corey, K.E.; Mendez-Navarro, J.; Barlow, L.L.; Patwardhan, V.; Zheng, H.; Kim, A.Y.; Lauer, G.M.; Chung, R.T. Acute hepatitis C infection lowers serum lipid levels. J. Viral Hepat. 2011, 18, e366–e371.
- Gastaminza, P.; Dryden, K.A.; Boyd, B.; Wood, M.R.; Law, M.; Yeager, M.; Chisari, F.V. Ultrastructural and biophysical characterization of hepatitis C virus particles produced in cell culture. J. Virol. 2010, 84, 10999–11009.
- Maillard, P.; Krawczynski, K.; Nitkiewicz, J.; Bronnert, C.; Sidorkiewicz, M.; Gounon, P.; Dubuisson, J.; Faure, G.; Crainic, R.; Budkowska, A. Nonenveloped nucleocapsids of hepatitis C virus in the serum of infected patients. J. Virol. 2001, 75, 8240–8250.
- Vercauteren, K.; Mesalam, A.A.; Leroux-Roels, G.; Meuleman, P. Impact of lipids and lipoproteins on hepatitis C virus infection and virus neutralization. World J. Gastroenterol. 2014, 20, 15975–15991.
- Lindenbach, B.D.; Rice, C.M. The ins and outs of hepatitis C virus entry and assembly. Nat. Rev. Microbiol. 2013, 11, 688–700.
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. EGFR and EphA2 are host factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595.
- Wang, C.; Gale, M.; Keller, B.C.; Huang, H.; Brown, M.S.; Goldstein, J.L.; Ye, J. Identification of FBL2 as a geranylgeranylated cellular protein required for hepatitis C virus RNA replication. Mol. Cell 2005, 18, 425–434.
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog. 2012, 8, e1003056.
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The lipid droplet is an important organelle for hepatitis C virus production. Nat. Cell Biol. 2007, 9, 1089–1097.
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 2003, 77, 5487–5492.
- Egger, D.; Wölk, B.; Gosert, R.; Bianchi, L.; Blum, H.E.; Moradpour, D.; Bienz, K. Expression of hepatitis C virus proteins induces distinct membrane alterations including a candidate viral replication complex. J. Virol. 2002, 76, 5974–5984.
- Mingorance, L.; Castro, V.; Ávila-Pérez, G.; Calvo, G.; Rodriguez, M.J.; Carrascosa, J.L.; Pérez-Del-Pulgar, S.; Forns, X.; Gastaminza, P. Host phosphatidic acid phosphatase lipin1 is rate limiting for functional hepatitis C virus replicase complex formation. PLoS Pathog. 2018, 14, e1007284.
- Stoeck, I.K.; Lee, J.Y.; Tabata, K.; Romero-Brey, I.; Paul, D.; Schult, P.; Lohmann, V.; Kaderali, L.; Bartenschlager, R. Hepatitis C Virus Replication Depends on Endosomal Cholesterol Homeostasis. J. Virol. 2018, 92.
- Lee, J.Y.; Cortese, M.; Haselmann, U.; Tabata, K.; Romero-Brey, I.; Funaya, C.; Schieber, N.L.; Qiang, Y.; Bartenschlager, M.; Kallis, S.; et al. Spatiotemporal Coupling of the Hepatitis C Virus Replication Cycle by Creating a Lipid Droplet- Proximal Membranous Replication Compartment. Cell Rep. 2019, 27, 3602–3617.