Genomic integrity is maintained by DNA repair and the DNA damage response (DDR). Defects in certain DNA repair genes give rise to many rare progressive neurodegenerative diseases (NDDs), such as ocular motor ataxia, Huntington disease (HD), and spinocerebellar ataxias (SCA). A possible causative role for DNA damage and DNA repair in the pathogenesis of common, late-onset NDDs is less well established, although DDR defects are emerging as possible culprits in diseases like AD, PD, and ALS. Whether targeting DNA repair or the DDR may be developed into therapeutic options against NDDs remains to be clarified.
- neurodegeneration
- iPSCs
- DNA damage response
- SOD
- organoids
- cGAS/STING
- autophagy
- cGAS-STING
1. Introduction
The basic unit of our body, the cell, constantly encounters stress in various forms. Even though the brain is generally protected from many environmental agents, some external and endogenous stress conditions damage the genetic material (DNA). Endogenous DNA damaging agents are by-products of normal cellular metabolism. As a consequence of a high rate of oxygen consumption and metabolic activity, brain cells sustain a high burden of reactive oxygen species (ROS) that attack the DNA by oxidizing its bases and backbone. Having the lowest oxidation potential, the DNA base guanine is highly susceptible to oxidation [1]. Thus, the major, and best studied, DNA oxidation product 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxoG) is among the most frequently used biomarkers for oxidative stress [2]. Hydrolytic deamination is another source of DNA damage under physiological conditions, leading to the loss of DNA bases and base modifications [3][4].
Moreover, many environmental agents associated with neurotoxicity and the development of neurodegenerative diseases (NDDs) [5], such as heavy metals (Pb, Cd, As, Hg, Cu, Zn and Fe) and pesticides (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), paraquat, dieldrin, rotenone), induce DNA damage via ROS formation. Rotenone and MPTP, for example, uncouple the mitochondrial electron transfer chain by inhibiting mitochondrial complex I [6]. There are two major strategies to protect DNA: antioxidant defense and DNA repair. There is ample evidence that the former is crucial for neuronal health, and several clinical trials are aiming to boost or stimulate antioxidant capacity.
Ageing is one of the main risk factors for most neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD) [7][8]. Hallmarks of ageing include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient-sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication [9]. Most of these biological processes may directly or indirectly contribute to NDDs. For instance, progressive damage to both nuclear DNA and mitochondrial DNA (mtDNA) is associated with aging. Damaged nuclear DNA can also lead to the activation of nucleus to mitochondria (NM) signaling. NM signaling is also associated with oxidative stress (through ROS generation and accumulation), which may ultimately contribute towards NDD [10]. Although oxidative DNA damage is believed to promote the aging process and contribute to the pathogenesis of several NDDs [11], the neuroprotective function of DNA repair is less clear. Proof of concept that DNA repair is important for brain health is provided by the existence of several rare NDDs caused by defects in DNA repair or DNA damage response (DDR) signalling, e.g., ocular motor apraxia, Huntington disease (HD) and certain cerebellar and spinocerebellar ataxias (SCA) [12].
In general, DDR signaling is activated in response to DNA damage. Central enzymes coordinating downstream DDR signaling include Ataxia Telangiectasia mutated (ATM), Ataxia Telangiectasia mutated and Rad3-related (ATR), and DNA-dependent protein kinase (DNA-PK). The DDR is highly complex and interconnected, and the precise pathways activated depend on the type of DNA damage, cell type and cell cycle stage. In general, the purpose of the DDR is threefold: (i) to detect and repair DNA damage, (ii) to coordinate the cells’ responses to DNA damage via activating a complex cellular cascade, and (iii) to induce cell cycle arrest or apoptosis if needed (we refer to recent reviews [13]). This orchestration is achieved by a DNA damage sensor, that, after activation, induce a signal transduction cascade with direct targets that promote several components of the cellular response to DNA damage, e.g., ATM phosphorylates H2A.X variant histone (gH2AX) which is important for transcription rewiring and the recruitment of repair proteins to chromatin. Of the many ATM targets, p53 is a master regulator of the cell cycle, which, when phosphorylated, leads to G1 arrest [14] and transcriptional activation of pro-apoptotic proteins [15].
2. DNA Damage Response and Repair Mechanisms
Mammalian cells are equipped with a highly elaborate DDR program that coordinates the cellular response to DNA damage [16]. Six major DNA repair mechanisms are integral to the DDR (Figure 1). Of these, direct reversal (DR), nucleotide excision repair (NER), base excision repair (BER), and mismatch repair (MMR) remove damaged and mismatched bases. Double-strand break repair (DSBR) pathways include non-homologous end joining (NHEJ) and homologous recombination (HR) [17]. NER is one of the most versatile DNA repair pathways and can handle a variety of lesions such as bulky DNA adducts formed by exposure to environmental genotoxic agents [18]. BER primarily corrects damaged DNA bases that do not create major structural distortions in DNA [19]. BER proteins are also important for the repair of single-strand breaks (SSBs). MMR corrects small insertion/deletion loops or mispaired nucleotides [20]. BER is considered to be the primary mechanism to counteract mitochondrial DNA damage [21]. The canonical NER, MMR and DSBR pathways do not operate in the mitochondria because many key enzymes are not localized in mitochondria. However, individual proteins are found in mitochondria, such as the NER proteins Cockayne Syndrome group B (CSB) and Xeroderma Pigmentosum Group D Protein (XPD) [22][23]. It remains to be demonstrated whether DNA repair pathways other than BER function in mitochondria, or other strategies are used to ensure mitochondrial DNA maintenance [24]. HR repairs double-strand breaks (DSBs) and inter-strand DNA crosslinks. Because HR requires a sister chromatid as a template for repair, it is restricted to S and G2 phases of the cell cycle, and is thus considered a “faithful DNA repair pathway”. NHEJ, in contrast, does not require homology and can directly seal blunt-ended breaks. Thus, NHEJ is active throughout the cell cycle [25][26]. As NHEJ does not utilize a template for repair and may involve end trimming to prepare ends for relegation [27], there is a risk that mutations are introduced. NHEJ is therefore referred to as “error-prone” [28]. Similar to nuclear DNA, mitochondrial DNA is also exposed to endogenous and exogenous agents.
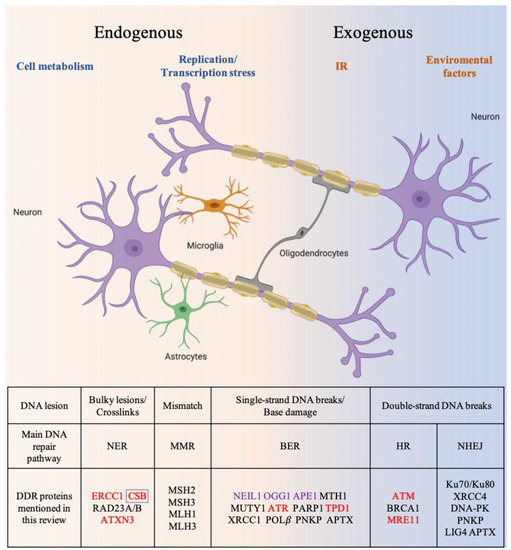
In addition to DNA repair, DDR involves the activation of signaling pathways that coordinate the cellular responses to DNA damage (see [30] or [31] for a recent review). ATM, ATR, and DNA-PK orchestrate the downstream DDR signaling pathways by phosphorylating hundreds of enzymes [13]. Both ATM and DNA-PK catalytic subunits (DNA-PKcs) are activated in response to DNA double-strand breaks (DSB) [13] through different DNA damage sensor complexes. After activation by the MRE11-RAD50-NBS1 (MRN) complex, ATM phosphorylates several downstream DDR proteins, as described above [32][33][34][35][36]. However, ATM also activates several proteins that function in other cellular pathways. For example, ATM is also a regulator of mitophagy, oxidative stress responses and insulin signaling [37][38]. DNA-PKcs is activated by the Ku70/Ku80-DNA complex and is required for NHEJ [13][39]. Through its participation in NHEJ, DNA-PKcs plays a role in lymphocyte and neuronal differentiation, and also functions in post-integrational DNA repair in the case of human immunodeficiency virus-1 (HIV-1) infection [39][40][41]. During lymphocyte development, DSBs are generated in immunoglobulin and T cell receptor loci to generate immune-receptor diversity by V(D)J and class-switch recombination. DNA-PK mediated NHEJ is required to repair these DSBs for joining V(D)J recombination intermediates [40]. NHEJ factors (including DNA-PK) also play important roles in repairing DSBs that arise during the differentiation of neural progenitor cells. Specific mutations in DNA-PKcs affect NHEJ repair, and thus could lead to profound neurological defects [41][42]. ATR is closely related to ATM and DNA-PKcs [13]. ATR is activated by long stretches of single-stranded DNA coated by Replication Protein A (RPA) and many types of genomic stress, including stalled replication fork and transcription. Thus, in addition to its role in activating DNA damage checkpoint, ATR also functions in unperturbed DNA replication [43].
3. NDDs related to the defective DNA repair mechanisms
Defects in DNA repair proteins cause several rare diseases with neurodegeneration as part of the clinical phenotype. These rare disorders establish the general concept that neurodegeneration may result from defects in DNA repair. For instance, mutations in APTX, PNKP or XRCC1 (all single-strand break repair (SSBR) genes) are linked to ocular motor apraxia [12] whereas defects in TDP1 (a SSBR gene) can lead to SCA with axonal neuropathy [44]. Similarly, mutations in the DSBR genes MRE11 and the main DNA damage sensors proteins, ATM and ATR, can lead to cerebellar ataxia [13][45].
Moreover, DNA repair is involved in regulating the length of CAG repeats in some trinucleotide repeat disorders such as HD and certain SCAs [46]. GWAS studies in 4000 HD patients have shown a number of variants in genes that encode DDR components, particularly in MMR genes [47]. Much remains to be clarified concerning the mechanisms involved, but MMR and BER proteins have been suggested to generate the nicked DNA intermediate that serves as a template for expansion [48][49]. This is supported by CAG repeat expansion being abrogated in MMR-defective (e.g. Msh2, Msh3, Mlh1 or Mlh3 knockout) or BER-defective (e.g. Ogg1 or Neil1 knockout) mouse models of HD. Attenuation of somatic or germline repeat expansion was accompanied by amelioration of the HD-like phenotypes in some cases [50], which further supports a direct role of DNA repair enzymes in regulating CAG repeat stability. Conversely, it has been proposed that CAG-repeat containing proteins, in which repeat expansion causes neurological disorders, function in DNA repair: Examples include HTT, the product of the huntingtin gene mutated in HD, which is recruited by ATM to sites of DNA damage [51]. Similarly, ATXN3, causing spinocerebellar ataxia 3 (SCA3), interacts with RAD23A/B which are important players in NER [51]. Mutant ATXN3 has also been found to sequester PNKP outside nucleus and thereby impair its ability to take part in DNA repair [52].
Recently, another indirect link between DNA repair and NDDs was shown in Spinal Motor Atrophy (SMA) disease, which is caused by deletion or mutation in survival of motor neuron1 (SMN1) gene [53]. The disease occurs in early childhood causing motor neuron degeneration leading to muscular atrophy, asymmetric limb paralysis and death [53]. In SMA patient primary fibroblasts and non-dividing SMA neurons, low level of SMN leads to DNA-PKcs deficiency, which impairs NHEJ causing accumulation of R-loops, DNA damage and neurodegeneration [54]. Conversely, increased gene expression of survival of motor neuron2 (SMN2, a second copy of SMN1) [54][55]. as well as SMN1 ectopic expression reduce R-loops, restore DNA-PKcs level and enhances NHEJ mediated DNA repair with reduced neuronal degeneration [54][55].
These and similar reports establish that DNA repair pathways may contribute directly or indirectly towards NDDs. A possible causative role for DNA damage and DNA repair in the pathogenesis of common, late onset NDDs is less well established, although DDR defects are emerging as possible culprits in diseases like AD, PD and ALS (Figure 2).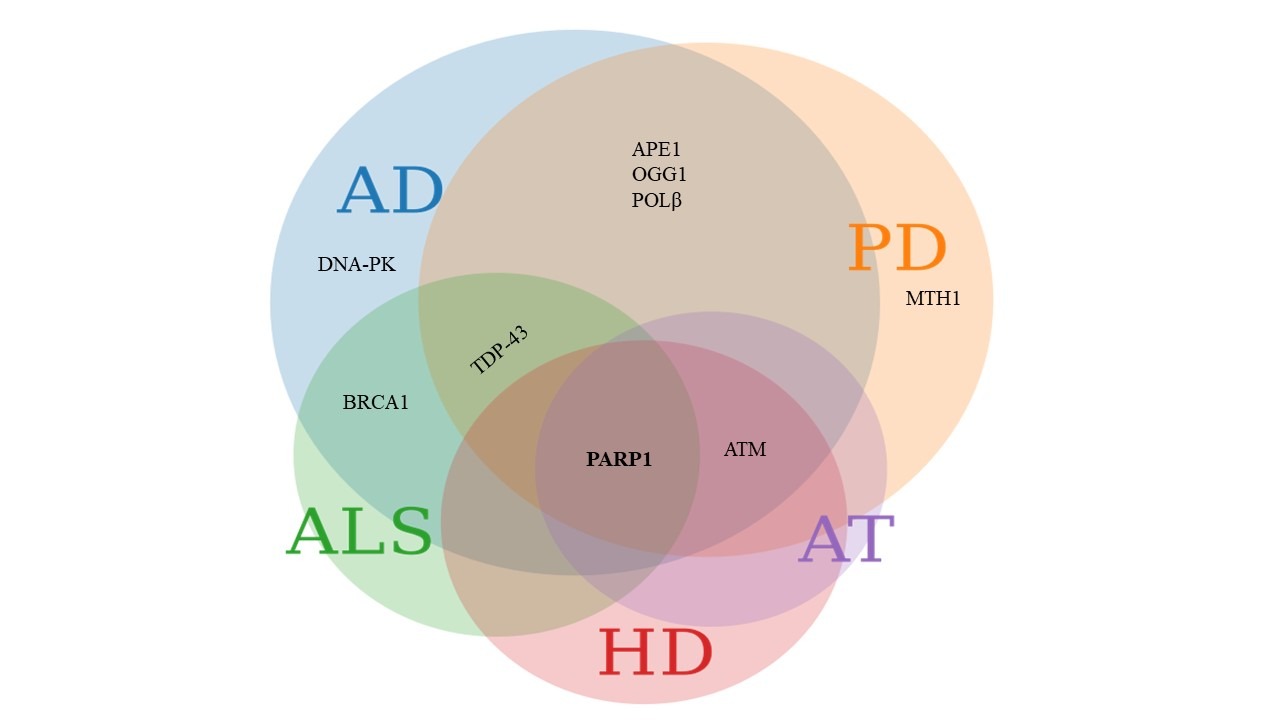
Figure 2. Common DDR proteins involved in various NDDs. The Venn diagram represents DDR proteins linked with various NDDs. Proteins involved in AD are depicted in blue circle, PD in orange, ALS in green, HD in pink and AT in purple. APE1, OGG1 and POLβ are involved in both AD and PD. Pathology related to the ALS associated protein TDP-43 is seen in both AD and PD. BRCA1 is involved in ALS and AD. ATM is dysregulated in AD, PD, HD and AT. Poly [ADP-ribose] polymerase 1 (PARP1) emerges as commonly implicated in AD, PD, ALS, HD and AT. The figure was created using meta chart software.
3.1. Alzheimer’s disease
Alzheimer’s disease (AD) is the most common age-related neurodegenerative disease responsible for 60-70% of dementia cases worldwide [56]. AD is characterized by progressive impairment in cognitive function, impaired decision-making ability, behavioral disturbances, and gradual memory loss leading to dementia [57]. The disease presents two major neuropathological hallmarks - extracellular Aβ, deposited as neuritic or amyloid plaques, and neurofibrillary tangles (NFT) comprising highly aggregated hyperphosphorylated TAU [57].
AD is considered as a multifactorial disease which involves complex interactions between intrinsic (ageing, genetics) and extrinsic (lifestyle, diet, environment) factors [58][59][60]. AD can be either familial, with mutations in APP, APOE, PSEN1 and PSEN2 genes [61][62][63] or sporadic which accounts for more than 90% of cases. Sporadic AD is a polygenic disease [64] with a strong contribution of APOE [65][66].
Several types of DNA damage, e.g. DSB, SSB and 8-oxoG, are detected in AD brains [67][68][69][70][71]. Reduced expression and activity of many DDR proteins (ATM, BRCA1 and DNA-PK) have been associated with AD pathology [72][73][74]. Similarly, expression of BER genes, like OGG1 and NEIL1, is reduced in AD [75][76][77]. Another BER gene, DNA Polymerase β (POLβ), whose expression is reduced during senescence and aging [78][79], is also suggested to play some role in AD, as reduction in DNA-gap filling activity in mild cognitive impairment (MCI) and AD brain was accompanied by decreased expression of POLβ [76]. Consistently, transgenic AD mice with genetic downregulation of Polβ (Polβ+/-) showed impaired memory and synaptic plasticity, increased neuronal death, and DNA damage accumulation [80]. All these studies indicate there is a correlation between AD and inefficient DNA repair, however further studies are required to establish a direct causal link between the two.
3.2 Parkinson´s Disease
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting 1% of the population over 60 years of age worldwide. The disease is characterized by asymmetrical bradykinesia, rigidity, resting tremor, and postural instability. The cardinal pathologic features of PD include progressive loss of dopaminergic neurons in the pars compacta of substantia nigra (SN), α-synuclein (α-SYN) aggregation, and Lewy body formation in the mid-brain region. Like AD, PD is a multifactorial disease caused by both environmental and genetic factors. Familial PD are associated with mutations in LRRK2, PARK7, PINK1, or SNCA [81][82].
Since oxidative stress and mitochondrial dysfunction are prominent features in PD, there has been a focus on studying the neuroprotective properties of mitochondrial DNA repair. Indeed, recent genetic data suggested that, on a pathway level, variants in genes involved in mtDNA maintenance may be enriched in PD patients [83]. The role for nuclear DNA repair is less well established, but elevated levels of 8-oxoG, SSBs and DSBs have been found in both PD post mortem human brains and in rat PD models [84][85][86]. Mice heterozygous for a deletion in the NER gene Ercc1, which only functions in nuclear DNA repair, had increased levels of phosphorylated α-SYN, γH2AX foci in the striatum, and developed severe PD-like pathology following MPTP administration [87]. These findings support that the nuclear DNA repair machinery play a role in PD pathology, but the mechanisms involved remain elusive.
The BER DNA glycosylases OGG1, MTH1 and MUTY1 are highly expressed in SN and associated dopaminergic neurons in PD brains [88][89][90]. Consistently, Ogg1 knock-out mice showed behavioral defects, and elevated 8-oxoG levels [91]. A direct, functional coupling between PD susceptibility genes and BER is suggested from the observation that Parkin ubiquitinates Apurinic/apyrimidinic endonuclease 1 (APE1) under stress. Moreover, mutations in PRKN abrogate APE1 ubiquitination resulting in overactivation of APE1 resulting in SSBs formation [92]. Elevated levels of AP sites were found in mtDNA in nigra l dopaminergic neurons from PD patients [93]. Polymorphisms in APE1 and OGG1 have been suggested to increase the risk of PD [94][95], but no individual single nucleotide polymorphism (SNP) in BER genes is consistently found to be associated with PD risk across cohorts [96][97]. However, elevated levels of pathological α-SYN correlated with increased PARP1 levels and PARylation in brain and cerebrospinal fluid of PD patients, supporting activation of cellular responses to SSBs. Interestingly, nuclear α-SYN may bind DNA and DDR proteins [98] and increased PARylation has been shown to promote pathological α-SYN aggregates [99]. Taken together, evidence of a direct functional coupling between DDR and α-SYN is emerging, suggesting a potential role for nuclear DDR in proteotoxicity in PD.
3.3 Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is an adult onset degenerative motor neuron disease, affecting the motor cortex, brain stem and spinal cord, leading to progressive paralysis and eventually death [100]. ALS is considered, primarily, a sporadic disease, but about 10% of ALS cases are familial [101]. More than 40 genes are currently linked to ALS, and the majority of cases, both familial (fALS) and sporadic ALS (sALS), are caused by mutations in four genes: C9ORF72, SOD1, TARDBP and FUS/TLS [102][103]. Over 30 years ago, a hypothesis suggesting the accumulation of abnormal DNA as the primary abnormality in ALS was brought forward [104] This has later been substantiated by many studies showing AP sites, DNA SSBs and 8-oxoG accumulation in diseased human motor neurons [105][106] further supporting that defective DDR is involved in ALS.
Defects in SOD1, the first identified ALS gene, which act as scavenger against free radicals, link oxidative damage with ALS [107]. Activation of several key proteins that function in sensing DNA damage, e.g. ATM, has been shown in human ALS motor neurons with SOD1 mutations, suggesting DNA damage accumulation in SOD1 deficient neurons [105]. Whole exome sequencing revealed NIMA-related kinase 1 (NEK1) as another ALS associated gene. NEK1 functions in cell-cycle checkpoint control [108] and contributes to DDR independent of ATM and ATR [109]. Human induced-pluripotent stem cells (iPSCs) and differentiated motor neurons carrying NEK1 mutations show dysregulation of the DDR machinery and increased DNA damage [110].
APE1 and OGG1 are upregulated in ALS brains [111] and spinal cord motor neurons in SOD1 transgenic mice, respectively, indicating increased DNA oxidative base damage [112]. Hypomethylation of the APE1 and OGG1 promoter regions have been recently described in ALS [105]. The authors speculated that hypomethylation might represent a compensatory upregulation of these BER genes in the vulnerable ALS neurons in order to cope with oxidative stress. Interestingly, two RNA/DNA binding proteins encoded by ALS pathogenic genes, TARDBP and FUS/TLS, function in DNA damage response. Mitra et al. showed DSBs accumulation and reduced recruitment of the XRCC4-XLF-DNA ligase 4 (LIG4) complex at DSB sites, thus indicating attenuated NHEJ, in TDP-43 (encoded by TARDBP) depleted motor neurons. Moreover, the authors demonstrated that TDP-43 acts as a scaffold facilitating recruitment of the XRCC4/LIG4 complex to DSBs [113]. Recent studies revealed that FUS is recruited to DSBs and interact directly with PARP1 [114][115]. Moreover, interaction between FUS and Histone deacetylase 1 (HDAC1) promotes NHEJ [116][117]. A non-canonical translation mechanism leads to production of five dipeptide repeat proteins (DPRs) from the hexanucleotide repeat expanded C9ORF72 gene. A recent study indicated that, among the five DPRs, proline-arginine (PR), glycine-arginine (GR), and glycine-alanine (GA) are the most neurotoxic and decrease the efficiency of NHEJ, single-strand annealing and microhomology-mediated end joining (MMEJ) [118]. Consistently, increased levels of several DDR markers (γH2AX, phosphorylated ATM, cleaved PARP1 and 53BP1) were observed in spinal cord motor neurons of C9ORF72 ALS patients [119].
Taken together, these studies suggest that several DNA repair pathways are implicated in NDDs pathogenesis, however the exact molecular mechanisms by which the responses to genomic stress contribute to neurodegeneration is still unclear.
4. Crosstalk between DDR and age-related neurodegenerative diseases
From the above it is clear that impaired DDR may contribute directly or indirectly towards various NDDs. Moreover, individual DDR proteins may be involved in more than one NDDs (Figure 2). Dysregulation, dysfunction, or inactivation of ATM is reported in AD and PD. Using a fly model, Petersen et al. showed that even a slight reduction in ATM kinase activity caused neurodegenerative features [120]. Reduced ATM levels and activity have been shown in hippocampal and frontal cortex neurons in AD brain as well as in AD transgenic mice [72]. Dysregulation of ATM signaling was also reported in HD where, contrary to AD or AT, increased or persistent activation of ATM signaling correlated with disease progression [121][122]. In a rodent PD model, α-synucleinopathy led to upregulation of γH2AX, 53BP1 and phosphorylation of ATM in neurons [87][123] suggesting that DSB might contribute to DA neurodegeneration in aging brain.
The DSB repair protein BRCA1 affects cognitive function in transgenic human amyloid precursor protein (hAPP) AD mice where shRNA mediated knockdown was accompanied by a reduction of memory and learning ability [73]. In post mortem AD brain, hypomethylation of the BRCA1 promoter region was accompanied by upregulation of expression and cytosolic mislocalization of BRCA1 [124]. In AD, expression of BRCA1 was increased, possibly as a consequence of oxidative DNA damage accumulating due to Aβ-induced ROS formation [125]. However, in this case, upregulation of BRCA1 did not translate to increased DDR capacity because BRCA1 remained dysfunctional and co-aggregated with TAU in a highly insoluble form, making it unavailable for DNA repair [124][126][127][128]. Hence, increased expression does not always lead to increased repair capacity but it is suggested that BRCA1 can function by a dual mechanism in AD: While it can cause abnormal ubiquitination and sub-cellular distribution of presenilin 1 (PS1) and thereby affect Aβ processing, it can also induce pro-apoptotic signaling [128]. Hence, also in AD the emerging evidence suggests a direct functional coupling between DDR and central pathogenic components. Whether this is also the case in ALS remains to be determined, but transcriptomic profiling shows that BRCA1 is highly expressed in the microglia of human ALS patients [129].
Mice deficient in the NHEJ repair enzymes LIG4, XRCC4 along with Ku70 and Ku80 show apoptosis of post mitotic neurons [130]. Abnormal expression of DNA-PK has been reported in AD. Reduced NHEJ activity in cortical neurons and cortical extracts from AD brains was ascribed to lower protein levels of DNA-PK and its regulatory subunit Ku80 [131][132]. Aβ aggregates inhibit DNA-PK activity in nerve growth factor-differentiated PC12 cells by reducing the expression of DNA-PKcs in a ROS dependent manner [133]. Aβ can enter the nucleus of PC12 cells and downregulate the expression of DNA-PKcs through a mechanism independent of oxidative stress, thus, indicating that Aβ itself may attenuate DNA-PKcs activity and hence reduce NHEJ capacity. As mentioned above, DNA-PK deficiency also leads to SMA [55].
PARP1 is activated by SSB to synthesize poly-ADP ribose (PAR) polymers at the damaged site [134][135][136][137][138][139]. Constitutive PARP1 activation can deplete intracellular NAD+ levels which can affect mitochondrial homeostasis, ROS production, DNA repair, and cell death [140][141][142]. A correlation between PARP1 activation and AD has been shown in AD human brains and AD mouse models [143][144][145][146]. Also, it has been shown that MPTP-treated mice exhibit PARP1 activation [147]. A mechanistic link between PARP1 activation and neurodegeneration was supported by Parp1−/−mice being resistant to the toxic effects of MPTP [147]. In ALS patients, PARP1-mediated, caspase independent programmed cell death of motor neurons through parthanatos was reported in the spinal cord [148][149][150]. Thus, PARP1 emerges as one of the central DDR proteins involved in most of the NDDs (Figure 2). The available evidence suggests that PARP1 mediated AD and PD pathologies could result from several cellular pathways: i) bioenergetic deficit via NAD+ depletion, ii) activation of apoptosis via interaction with tumor suppressor and apoptotic genes like TP53 and BCL2; iii) induction of parthanatos; and iv) transcription rewiring via modulation of transcriptional factors [145]. A causal role for PARP1 activation in NDDs pathogenesis is further substantiated by studies where PARP1 inhibition and NAD+-augmentation prevented neuron degeneration in PD animal models [99][151]. Based on this Nicotinamide riboside supplementation was proposed as a possible therapeutic intervention for NDDs [151].
5. Crosstalk between DDR and other cellular processes known to be disturbed in NDDs
From the above, it is apparent that defects in DNA repair proteins may contribute towards the initiation and progression of more than one NDD. In this section, we will describe some of the unifying mechanisms observed in various NDDs and will provide the evidence for their crosstalk with the DDR machinery (Figure 3).
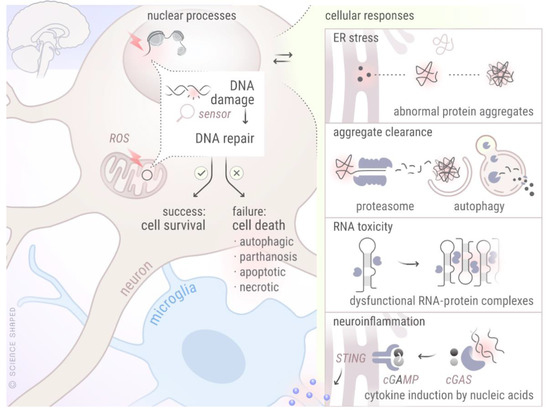
Figure 3. Crosstalk between different cellular mechanisms involved in various NDDs. The figure represents crosstalk between DDR and other cellular processes known to be perturbed in various NDDs. Created by Ellen Tenstad @ScienceShaped.
5.1. DDR and Autophagy
Inefficient aggregate clearance indicating defective autophagy is one of the common features of NDDs. During AD pathogenesis, both impaired autophagosome synthesis and reduced clearance of autophagic substrates have been shown [152][153][154]. Accumulation of α-SYN in PD indicates dysfunctional chaperone mediated autophagy (CMA), a major contributor for the autophagic degradation of α-SYN [155]. Mutations in many autophagy receptors, including SQSTM1/p62 [155][155], OPTN, and UBQLN2, have been associated with the pathogenesis of ALS [156][157][158]. Associations between DDR and autophagy as a direct cause of NDDs are not yet well-established, although literature suggests indirect connections between the DNA repair machinery and autophagy through various, non-exclusive routes [159][160][161]. ATM activation can cause autophagy induction through 5' AMP-activated protein kinase (AMPK) in response to ROS [162][163]. While Atm-/- neurons show abnormal autophagy, ATM itself is processed through autophagic degradation [164]. Parp1 knockout mice subjected to acute starvation displayed deficient liver autophagy, implying a physiological role for PARP1 in starvation-induced autophagy [165]. The link between DDR and autophagy seems bidirectional as knocking out BECLIN1, a protein involved in autophagy induction, reduced activity and levels of crucial HR and NHEJ proteins, and also attenuated the formation of DNA-PK complexes [166][167]. SQSTM1/p62 has been shown to regulate the ratio between HR and NHEJ by promoting the latter upon radiation [168]. CMA regulates CHK1 levels and prevents hyperphosphorylation and destabilization of the MRN complex [169]. The translation product of ultraviolet irradiation resistance-associated gene (UVRAG), an important player in autophagy initiation, patrols DSB repair activity through direct binding to DNA-PK in NHEJ [170]. Interestingly, DNA damage could induce mitophagy, which may play a role to protect cells against DNA-damaged induced cellular death [171]. Although not explored for DDR, our group recently showed induction of endoplasmic stress (ER stress) mediated pro-survival autophagy after radiation induced ROS induction [172].
5.2. DDR and neuroinflammation
Neuroinflammation is another common phenomenon noticed in different NDDs. A vicious cycle is believed to operate involving neuroinflammation, oxidative stress, and neurodegeneration [173][174]. Inflammation can itself induce DNA damage through the generation of ROS and reactive nitrogen species (RNS). Inflammation increases, not only, mutagenic DNA lesions, such as 8-nitroguanine and 8-oxoG, but also negatively impacts on DNA repair capacity by inhibiting many important DNA repair enzymes [175][176]. The seminal discovery of cyclic GMP-AMP synthase (cGAS) as the mammalian cytosolic DNA sensor by Sun et al. has established direct links between damaged DNA and neuroinflammation [177]. Upon binding to cytosolic DNA, cGAS converts ATP and GTP to cGAMP which binds and activates the endoplasmic reticulum protein stimulator of interferon genes (STING), finally inducing the production of type I interferons through the transcription factors NF-kB and IRF3 [178]. Activation of cGAS-STING has been seen in several NDDs: In an Atm-deficient rat model with neuroinflammatory and neurodegenerative phenotypes, an accumulation of cytosolic DNA leading to enhanced levels of phosphorylated TBK1 (indicative of STING activation) was observed in various cell types including microglia [179]. Xu et al. demonstrated that cGAMP can suppress AD by elevating TREM2 levels [180]. In iPSC derived ALS patient motor neurons, TDP-43 triggered release of mtDNA, leading to cGAS-STING activation and neuroinflammation has been shown [181]. Induction of IL6 in response to circulating cell-free mtDNA has been reported in Parkinson patient’s serum [182]. Hence, cGAS-STING mediated inflammation emerges as another common characteristic of NDDs.
In summary, individual DDR protein can be involved in more than one NDDs (Figure 2), and can induce neurodegenerative features through their interaction with various cellular processes (Figure 3).
6. Model systems to study NDDs and therapeutics
Although post mortem brain tissue is useful to map major structural or molecular differences in DDR markers in late stages of NDDs, they have limited power to explain disease mechanisms. Thus, animal models remain essential tools to study mechanisms driving neurodegeneration in a life-course perspective. A selection of studies linking DDR to NDDs using various model systems discussed above or suggesting appropriate models to study this crosstalk is summarized in Table 1. As is evident from the above discussion, rodents remain the most extensively used model animals. For AD, for example, there are more than a hundred different rodent models (https://www.alzforum.org/research-models/search). However, no model recapitulates all cardinal phenotypes seen in the human disease, illustrating the need for better models to model human NDDs.
Table 1. Different model systems to study NDDs and the involvement of DDR. The table shows various model systems that have been used to study the role of DDR in various NDDs.
|
Model |
DDR Genes |
NDD |
References |
|
|
PARP1 |
AD |
[146] |
|
TARDBP |
ALS |
[183] |
|
|
C. elegans |
PARP1 |
AT |
[140] |
|
|
XPA |
XP |
[184] |
|
|
CSB |
CS |
[184] |
|
|
ATM |
AT |
[184] |
|
D. melanogaster |
ATM |
AT |
[72] |
|
AD |
[120] |
||
|
HD |
[122] |
||
|
D. rerio |
C9ORF72 |
ALS |
[185] |
|
ATM |
AT |
[186] |
|
|
M. musculus |
ATM |
HD |
[122] |
|
AD |
[72] |
||
|
BRCA1 |
ALS |
[129] |
|
|
AD |
[73] |
||
|
POLβ |
AD |
[80] |
|
|
OGG1 |
PD |
[91] |
|
|
ALS |
[112] |
||
|
ERCC1 |
PD |
[87] |
|
|
PARP1 |
AD |
[145] |
|
|
PD |
[147] |
||
|
ALS |
[150] |
||
|
R. norvegicus |
ATM |
PD |
[123] |
|
iPSCs |
BRCA1 |
AD |
[128] |
|
NEK1 |
ALS |
[110] |
Invertebrate animals are relatively inexpensive to maintain, have short lifespan and reproduction cycle. These properties make non-vertebrate models amenable for drug and genetic screening. The nematode Caenorhabditis elegans (C. elegans) is an example of an animal where the transparent body allows monitoring neurodegeneration in vivo. Humanized animal models are available in which transgenic animals express human α-SYN [187][188], Aβ1-42, TAU [189][190], TDP-43 and SOD1[191][192][193]. The existence of behavioral assays makes it possible to study viability and functionality from individual neurons during a natural life course. Hence, simple animals offer excellent opportunities to study the combined effects of genetic and environmental risk factors in conjunction with aging, the most important risk factor, for NDD. With respect to the contribution of DDR, C. elegans has less redundancy of DNA repair genes making it a great tool to define whether and how DNA repair proteins affect neurodegeneration. A limitation of C. elegans is that it does not have a brain. Thus, vertebrate models are needed. Of these, Danio rerio (D. rerio) is increasingly popular as it has vasculature and a brain separated by a blood-brain barrier [194][195][196].
The development of human iPSC technology has opened the doors to modeling complex brain diseases. Established protocols are available to generate various brain cell types from patient derived iPSCs in two dimensional (2D) monolayer cultures and use these to model AD [197][198], ALS [199][200][201] and PD [202][203]. The possibility of introducing genome editing in iPSCs (e.g. via CRISPR/CAS9) further increases the potential of these cells to study the specific roles of individual genes. In addition, the role of other brain cell types (including glial cells, also derived from iPSCs) can be studied in recently developed co-culture systems [204]. Still, the 2D culture systems do not recapitulate the complexity of the brain, and phenotypes such as extracellular protein aggregation are difficult to observe. Thus, 2D iPSCs are not the most preferred choice to model NDDs. Self-organization of embryoid bodies from iPSCs, so-called 3D-organoid models may allow us to study cellular development and inter-cellular interactions within a 3D human brain microenvironment [205]. Advances in 3D-culture systems have led to the formation of multiple brain cell types and specific brain regions in these organoids (mini-brains) [206]. Forebrain cortical organoids, mid-brain organoids, and motor neuron spheroids derived from AD and HD, PD and ALS patient iPSCs, respectively, are able to show the cardinal tissue phenotypes and can, thus, be very useful for understanding these complex NDDs (Figure 4) [207][208][209][210][211][212][213].
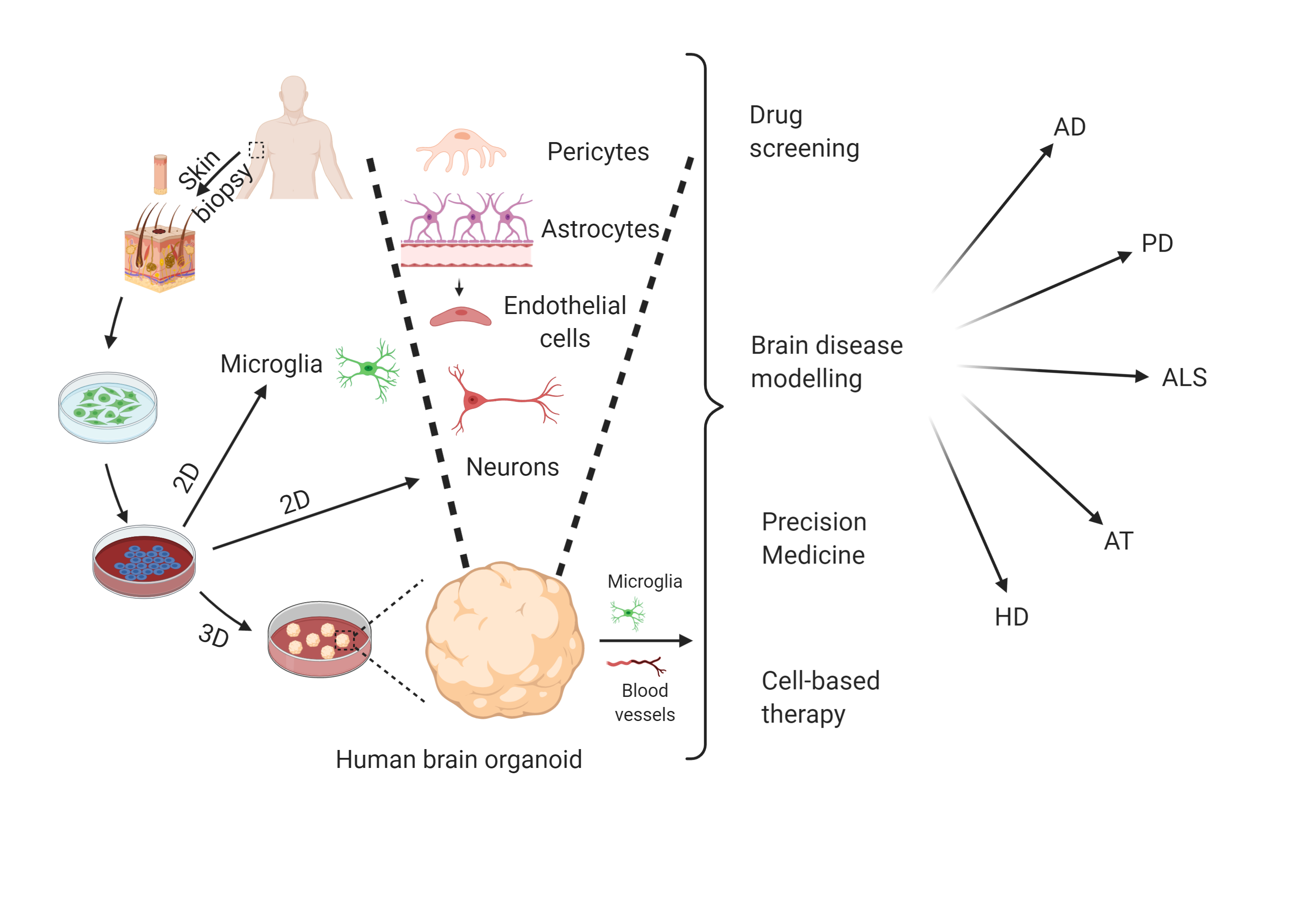
Figure 4. Potential of 2D iPSC and 3D-organoids to model NDDs. Skin fibroblasts derived from NDD patients can be reprogrammed into iPSCs. These iPSCs can be differentiated into various brain cell types including different neuron types, microglia etc. iPSC-derived 3D-organoids contain most of the brain cell types (except microglia and vasculature, which can be co-cultured with 3D-organoids) and are able to show major disease phenotypes. Various applications of these model systems have also been shown. Created with BioRender.com.
7. Conclusion
To summarize, both antioxidant defense and DNA repair prevent accumulation of DNA damage and thereby counteract development of NDDs. One example for antioxidant defense is the scavenger superoxide dismutase1 (SOD1) that protects neurons against amyloid β (Aβ) mediated neurotoxicity [214]. Both overexpression of wild type SOD1 and mutations in SOD1 are associated with several age-related NDDs [11][215][216]. In fact, several clinical trials are targeting wild type SOD1 or mutant SOD1 to prevent aggregation and misfolding in NDDs and an antisense oligonucleotide is now in phase III clinical trial (NCT02623699). A small molecule, Arimoclomol, that promotes proper folding of SOD1 in the ER [11], is in phase III trial (NCT03491462). Whether targeting DNA repair or the DDR may be developed into therapeutic options against NDDs remain to be clarified. However, the emergence of PARP1 activation by SSB as a common finding in NDDs, and the availability of PARP1 inhibitors suggests that this might be a path to pursue. However, directly targeting PARP1 carries risks of deleterious effects on other cells and tissue systems. One consequence of PARP1 overactivation is depletion of cellular NAD+, which is a substrate of PARP1. In our previous work we showed that boosting NAD+ levels is a promising strategy to stimulate DNA repair and prevent neurodegeneration in rare DNA repair disease [140][146][184]. To further explore the therapeutic potential of DDR, cross-species studies where different model systems are combined to recapitulate different aspects of these complex diseases can be used. iPSC-derived 3D-organoid models emerge as promising model systems that can capture the complexity of the DDR and its crosstalk with other cellular processes important to maintain neuronal health.
References
- Delaney, S.; Jarem, D.A.; Volle, C.B.; Yennie, C.J. Chemical and biological consequences of oxidatively damaged guanine in DNA. Free Radic Res 2012, 46, 420-441, doi:10.3109/10715762.2011.653968.
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J Clin Invest 2012, 122, 4344-4361, doi:10.1172/JCI65053.
- Krokan, H.E.; Drablos, F.; Slupphaug, G. Uracil in DNA--occurrence, consequences and repair. Oncogene 2002, 21, 8935-8948, doi:10.1038/sj.onc.1205996.
- Lindahl, T.; Nyberg, B. Rate of depurination of native deoxyribonucleic acid. Biochemistry 1972, 11, 3610-3618, doi:10.1021/bi00769a018.
- Chin-Chan, M.; Navarro-Yepes, J.; Quintanilla-Vega, B. Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front Cell Neurosci 2015, 9, 124, doi:10.3389/fncel.2015.00124.
- Ramachandiran, S.; Hansen, J.M.; Jones, D.P.; Richardson, J.R.; Miller, G.W. Divergent mechanisms of paraquat, MPP+, and rotenone toxicity: oxidation of thioredoxin and caspase-3 activation. Toxicol Sci 2007, 95, 163-171, doi:10.1093/toxsci/kfl125.
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol 2019, 15, 565-581, doi:10.1038/s41582-019-0244-7.
- Wyss-Coray, T. Ageing, neurodegeneration and brain rejuvenation. Nature 2016, 539, 180-186, doi:10.1038/nature20411.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194-1217, doi:10.1016/j.cell.2013.05.039.
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 2016, 17, 308-321, doi:10.1038/nrm.2016.14.
- Trist, B.G.; Davies, K.M.; Cottam, V.; Genoud, S.; Ortega, R.; Roudeau, S.; Carmona, A.; De Silva, K.; Wasinger, V.; Lewis, S.J.G., et al. Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson's disease brain. Acta Neuropathol 2017, 134, 113-127, doi:10.1007/s00401-017-1726-6.
- Rulten, S.L.; Caldecott, K.W. DNA strand break repair and neurodegeneration. DNA Repair (Amst) 2013, 12, 558-567, doi:10.1016/j.dnarep.2013.04.008.
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol Cell 2017, 66, 801-817, doi:10.1016/j.molcel.2017.05.015.
- Kang, J.; Ferguson, D.; Song, H.; Bassing, C.; Eckersdorff, M.; Alt, F.W.; Xu, Y. Functional interaction of H2AX, NBS1, and p53 in ATM-dependent DNA damage responses and tumor suppression. Mol Cell Biol 2005, 25, 661-670, doi:10.1128/MCB.25.2.661-670.2005.
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 2018, 25, 104-113, doi:10.1038/cdd.2017.169.
- Zhou, B.B.; Elledge, S.J. The DNA damage response: putting checkpoints in perspective. Nature 2000, 408, 433-439, doi:10.1038/35044005.
- Yu, Z.; Chen, J.; Ford, B.N.; Brackley, M.E.; Glickman, B.W. Human DNA repair systems: an overview. Environ Mol Mutagen 1999, 33, 3-20, doi:10.1002/(sici)1098-2280(1999)33:1<3::aid-em2>3.0.co;2-l.
- Nouspikel, T. DNA repair in mammalian cells : Nucleotide excision repair: variations on versatility. Cell Mol Life Sci 2009, 66, 994-1009, doi:10.1007/s00018-009-8737-y.
- Nilsen, H.; Krokan, H.E. Base excision repair in a network of defence and tolerance. Carcinogenesis 2001, 22, 987-998, doi:10.1093/carcin/22.7.987.
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366-374, doi:10.1038/35077232.
- Fontana, G.A.; Gahlon, H.L. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res 2020, 48, 11244-11258, doi:10.1093/nar/gkaa804.
- Liu, J.; Fang, H.; Chi, Z.; Wu, Z.; Wei, D.; Mo, D.; Niu, K.; Balajee, A.S.; Hei, T.K.; Nie, L., et al. XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucleic Acids Res 2015, 43, 5476-5488, doi:10.1093/nar/gkv472.
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; de Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M., 3rd; Stevnsner, T., et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J 2010, 24, 2334-2346, doi:10.1096/fj.09-147991.
- Zhao, L.; Sumberaz, P. Mitochondrial DNA Damage: Prevalence, Biological Consequence, and Emerging Pathways. Chem Res Toxicol 2020, 33, 2491-2502, doi:10.1021/acs.chemrestox.0c00083.
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071-1078, doi:10.1038/nature08467.
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 2008, 7, 1765-1771, doi:10.1016/j.dnarep.2008.06.018.
- Brandsma, I.; Gent, D.C. Pathway choice in DNA double strand break repair: observations of a balancing act. Genome Integr 2012, 3, 9, doi:10.1186/2041-9414-3-9.
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J Biol Chem 2018, 293, 10502-10511, doi:10.1074/jbc.TM118.000371.
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat Cell Biol 2011, 13, 1161-1169, doi:10.1038/ncb2344.
- Nair, N.; Shoaib, M.; Sorensen, C.S. Chromatin Dynamics in Genome Stability: Roles in Suppressing Endogenous DNA Damage and Facilitating DNA Repair. Int J Mol Sci 2017, 18, doi:10.3390/ijms18071486.
- Lavin, M.F. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol 2008, 9, 759-769, doi:10.1038/nrm2514.
- Shiloh, Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 2003, 3, 155-168, doi:10.1038/nrc1011.
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Mol Cell 2010, 40, 179-204, doi:10.1016/j.molcel.2010.09.019.
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y., et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674-1677, doi:10.1126/science.281.5383.1674.
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359-370, doi:10.1016/s0092-8674(03)00566-x.
- Ditch, S.; Paull, T.T. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 2012, 37, 15-22, doi:10.1016/j.tibs.2011.10.002.
- Pizzamiglio, L.; Focchi, E.; Antonucci, F. ATM Protein Kinase: Old and New Implications in Neuronal Pathways and Brain Circuitry. Cells 2020, 9, doi:10.3390/cells9091969.
- Anisenko, A.; Kan, M.; Shadrina, O.; Brattseva, A.; Gottikh, M. Phosphorylation Targets of DNA-PK and Their Role in HIV-1 Replication. Cells 2020, 9, doi:10.3390/cells9081907.
- Alt, F.W.; Zhang, Y.; Meng, F.L.; Guo, C.; Schwer, B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013, 152, 417-429, doi:10.1016/j.cell.2013.01.007.
- Woodbine, L.; Neal, J.A.; Sasi, N.K.; Shimada, M.; Deem, K.; Coleman, H.; Dobyns, W.B.; Ogi, T.; Meek, K.; Davies, E.G., et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J Clin Invest 2013, 123, 2969-2980, doi:10.1172/JCI67349.
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X., et al. Differential phosphorylation of DNA-PKcs regulates the interplay between end-processing and end-ligation during nonhomologous end-joining. Mol Cell 2015, 58, 172-185, doi:10.1016/j.molcel.2015.02.024.
- Brown, E.J.; Baltimore, D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003, 17, 615-628, doi:10.1101/gad.1067403.
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 2017, 58, 235-263, doi:10.1002/em.22087.
- Kulkarni, A.; Wilson, D.M., 3rd. The involvement of DNA-damage and -repair defects in neurological dysfunction. Am J Hum Genet 2008, 82, 539-566, doi:10.1016/j.ajhg.2008.01.009.
- Coon, E.A.; Benarroch, E.E. DNA damage response: Selected review and neurologic implications. Neurology 2018, 90, 367-376, doi:10.1212/WNL.0000000000004989.
- Orr, H.T.; Zoghbi, H.Y. Trinucleotide repeat disorders. Annu Rev Neurosci 2007, 30, 575-621, doi:10.1146/annurev.neuro.29.051605.113042.
- Genetic Modifiers of Huntington's Disease, C. Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease. Cell 2015, 162, 516-526, doi:10.1016/j.cell.2015.07.003.
- McMurray, C.T. Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 2010, 11, 786-799, doi:10.1038/nrg2828.
- Panigrahi, G.B.; Lau, R.; Montgomery, S.E.; Leonard, M.R.; Pearson, C.E. Slipped (CTG)*(CAG) repeats can be correctly repaired, escape repair or undergo error-prone repair. Nat Struct Mol Biol 2005, 12, 654-662, doi:10.1038/nsmb959.
- Massey, T.H.; Jones, L. The central role of DNA damage and repair in CAG repeat diseases. Dis Model Mech 2018, 11, doi:10.1242/dmm.031930.
- Maiuri, T.; Mocle, A.J.; Hung, C.L.; Xia, J.; van Roon-Mom, W.M.; Truant, R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet 2017, 26, 395-406, doi:10.1093/hmg/ddw395.
- Gao, R.; Liu, Y.; Silva-Fernandes, A.; Fang, X.; Paulucci-Holthauzen, A.; Chatterjee, A.; Zhang, H.L.; Matsuura, T.; Choudhary, S.; Ashizawa, T., et al. Inactivation of PNKP by mutant ATXN3 triggers apoptosis by activating the DNA damage-response pathway in SCA3. PLoS Genet 2015, 11, e1004834, doi:10.1371/journal.pgen.1004834.
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Benichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155-165, doi:10.1016/0092-8674(95)90460-3.
- Kannan, A.; Bhatia, K.; Branzei, D.; Gangwani, L. Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res 2018, 46, 8326-8346, doi:10.1093/nar/gky641.
- Kannan, A.; Jiang, X.; He, L.; Ahmad, S.; Gangwani, L. ZPR1 prevents R-loop accumulation, upregulates SMN2 expression and rescues spinal muscular atrophy. Brain 2020, 143, 69-93, doi:10.1093/brain/awz373.
- Barker, W.W.; Luis, C.A.; Kashuba, A.; Luis, M.; Harwood, D.G.; Loewenstein, D.; Waters, C.; Jimison, P.; Shepherd, E.; Sevush, S., et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord 2002, 16, 203-212, doi:10.1097/00002093-200210000-00001.
- Griffin, W.S. Inflammation and neurodegenerative diseases. Am J Clin Nutr 2006, 83, 470S-474S, doi:10.1093/ajcn/83.2.470S.
- Castellani, R.J.; Rolston, R.K.; Smith, M.A. Alzheimer disease. Dis Mon 2010, 56, 484-546, doi:10.1016/j.disamonth.2010.06.001.
- Reitz, C.; Mayeux, R. Alzheimer disease: epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem Pharmacol 2014, 88, 640-651, doi:10.1016/j.bcp.2013.12.024.
- Dosunmu, R.; Wu, J.; Basha, M.R.; Zawia, N.H. Environmental and dietary risk factors in Alzheimer's disease. Expert Rev Neurother 2007, 7, 887-900, doi:10.1586/14737175.7.7.887.
- Kang, J.; Lemaire, H.G.; Unterbeck, A.; Salbaum, J.M.; Masters, C.L.; Grzeschik, K.H.; Multhaup, G.; Beyreuther, K.; Muller-Hill, B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature 1987, 325, 733-736, doi:10.1038/325733a0.
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K., et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995, 375, 754-760, doi:10.1038/375754a0.
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K., et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 1995, 269, 973-977, doi:10.1126/science.7638622.
- Wingo, T.S.; Lah, J.J.; Levey, A.I.; Cutler, D.J. Autosomal recessive causes likely in early-onset Alzheimer disease. Arch Neurol 2012, 69, 59-64, doi:10.1001/archneurol.2011.221.
- Bertram, L.; Tanzi, R.E. The genetic epidemiology of neurodegenerative disease. J Clin Invest 2005, 115, 1449-1457, doi:10.1172/JCI24761.
- Raber, J.; Huang, Y.; Ashford, J.W. ApoE genotype accounts for the vast majority of AD risk and AD pathology. Neurobiol Aging 2004, 25, 641-650, doi:10.1016/j.neurobiolaging.2003.12.023.
- Adamec, E.; Vonsattel, J.P.; Nixon, R.A. DNA strand breaks in Alzheimer's disease. Brain Res 1999, 849, 67-77, doi:10.1016/s0006-8993(99)02004-1.
- Myung, N.H.; Zhu, X.; Kruman, II; Castellani, R.J.; Petersen, R.B.; Siedlak, S.L.; Perry, G.; Smith, M.A.; Lee, H.G. Evidence of DNA damage in Alzheimer disease: phosphorylation of histone H2AX in astrocytes. Age (Dordr) 2008, 30, 209-215, doi:10.1007/s11357-008-9050-7.
- Shanbhag, N.M.; Evans, M.D.; Mao, W.; Nana, A.L.; Seeley, W.W.; Adame, A.; Rissman, R.A.; Masliah, E.; Mucke, L. Early neuronal accumulation of DNA double strand breaks in Alzheimer's disease. Acta Neuropathol Commun 2019, 7, 77, doi:10.1186/s40478-019-0723-5.
- Gabbita, S.P.; Lovell, M.A.; Markesbery, W.R. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem 1998, 71, 2034-2040, doi:10.1046/j.1471-4159.1998.71052034.x.
- Lyras, L.; Cairns, N.J.; Jenner, A.; Jenner, P.; Halliwell, B. An assessment of oxidative damage to proteins, lipids, and DNA in brain from patients with Alzheimer's disease. J Neurochem 1997, 68, 2061-2069, doi:10.1046/j.1471-4159.1997.68052061.x.
- Shen, X.; Chen, J.; Li, J.; Kofler, J.; Herrup, K. Neurons in Vulnerable Regions of the Alzheimer's Disease Brain Display Reduced ATM Signaling. eNeuro 2016, 3, doi:10.1523/ENEURO.0124-15.2016.
- Suberbielle, E.; Djukic, B.; Evans, M.; Kim, D.H.; Taneja, P.; Wang, X.; Finucane, M.; Knox, J.; Ho, K.; Devidze, N., et al. DNA repair factor BRCA1 depletion occurs in Alzheimer brains and impairs cognitive function in mice. Nat Commun 2015, 6, 8897, doi:10.1038/ncomms9897.
- Kanungo, J. DNA-PK Deficiency in Alzheimer's Disease. J Neurol Neuromedicine 2016, 1, 17-22, doi:10.29245/2572.942x/2016/3.1016.
- Lovell, M.A.; Xie, C.; Markesbery, W.R. Decreased base excision repair and increased helicase activity in Alzheimer's disease brain. Brain Res 2000, 855, 116-123, doi:10.1016/s0006-8993(99)02335-5.
- Weissman, L.; Jo, D.G.; Sorensen, M.M.; de Souza-Pinto, N.C.; Markesbery, W.R.; Mattson, M.P.; Bohr, V.A. Defective DNA base excision repair in brain from individuals with Alzheimer's disease and amnestic mild cognitive impairment. Nucleic Acids Res 2007, 35, 5545-5555, doi:10.1093/nar/gkm605.
- Canugovi, C.; Shamanna, R.A.; Croteau, D.L.; Bohr, V.A. Base excision DNA repair levels in mitochondrial lysates of Alzheimer's disease. Neurobiol Aging 2014, 35, 1293-1300, doi:10.1016/j.neurobiolaging.2014.01.004.
- Rao, K.S.; Annapurna, V.V.; Raji, N.S. DNA polymerase-beta may be the main player for defective DNA repair in aging rat neurons. Ann N Y Acad Sci 2001, 928, 113-120, doi:10.1111/j.1749-6632.2001.tb05641.x.
- Ahmed, A.A.; Smoczer, C.; Pace, B.; Patterson, D.; Cress Cabelof, D. Loss of DNA polymerase beta induces cellular senescence. Environ Mol Mutagen 2018, 59, 603-612, doi:10.1002/em.22206.
- Sykora, P.; Misiak, M.; Wang, Y.; Ghosh, S.; Leandro, G.S.; Liu, D.; Tian, J.; Baptiste, B.A.; Cong, W.N.; Brenerman, B.M., et al. DNA polymerase beta deficiency leads to neurodegeneration and exacerbates Alzheimer disease phenotypes. Nucleic Acids Res 2015, 43, 943-959, doi:10.1093/nar/gku1356.
- Gkikas, I.; Palikaras, K.; Tavernarakis, N. The Role of Mitophagy in Innate Immunity. Front Immunol 2018, 9, 1283, doi:10.3389/fimmu.2018.01283.
- Tan, M.M.X.; Malek, N.; Lawton, M.A.; Hubbard, L.; Pittman, A.M.; Joseph, T.; Hehir, J.; Swallow, D.M.A.; Grosset, K.A.; Marrinan, S.L., et al. Genetic analysis of Mendelian mutations in a large UK population-based Parkinson's disease study. Brain 2019, 142, 2828-2844, doi:10.1093/brain/awz191.
- Gaare, J.J.; Nido, G.; Dolle, C.; Sztromwasser, P.; Alves, G.; Tysnes, O.B.; Haugarvoll, K.; Tzoulis, C. Meta-analysis of whole-exome sequencing data from two independent cohorts finds no evidence for rare variant enrichment in Parkinson disease associated loci. PLoS One 2020, 15, e0239824, doi:10.1371/journal.pone.0239824.
- Alam, Z.I.; Jenner, A.; Daniel, S.E.; Lees, A.J.; Cairns, N.; Marsden, C.D.; Jenner, P.; Halliwell, B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem 1997, 69, 1196-1203, doi:10.1046/j.1471-4159.1997.69031196.x.
- Hegde, M.L.; Gupta, V.B.; Anitha, M.; Harikrishna, T.; Shankar, S.K.; Muthane, U.; Subba Rao, K.; Jagannatha Rao, K.S. Studies on genomic DNA topology and stability in brain regions of Parkinson's disease. Arch Biochem Biophys 2006, 449, 143-156, doi:10.1016/j.abb.2006.02.018.
- Yasuhara, T.; Hara, K.; Sethi, K.D.; Morgan, J.C.; Borlongan, C.V. Increased 8-OHdG levels in the urine, serum, and substantia nigra of hemiparkinsonian rats. Brain Res 2007, 1133, 49-52, doi:10.1016/j.brainres.2006.11.072.
- Sepe, S.; Milanese, C.; Gabriels, S.; Derks, K.W.; Payan-Gomez, C.; van, I.W.F.; Rijksen, Y.M.; Nigg, A.L.; Moreno, S.; Cerri, S., et al. Inefficient DNA Repair Is an Aging-Related Modifier of Parkinson's Disease. Cell Rep 2016, 15, 1866-1875, doi:10.1016/j.celrep.2016.04.071.
- Arai, T.; Fukae, J.; Hatano, T.; Kubo, S.; Ohtsubo, T.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Up-regulation of hMUTYH, a DNA repair enzyme, in the mitochondria of substantia nigra in Parkinson's disease. Acta Neuropathol 2006, 112, 139-145, doi:10.1007/s00401-006-0081-9.
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson's disease. J Neurosci Res 2007, 85, 919-934, doi:10.1002/jnr.21191.
- Fukae, J.; Takanashi, M.; Kubo, S.; Nishioka, K.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson's disease and related neurodegenerative disorders. Acta Neuropathol 2005, 109, 256-262, doi:10.1007/s00401-004-0937-9.
- Cardozo-Pelaez, F.; Sanchez-Contreras, M.; Nevin, A.B. Ogg1 null mice exhibit age-associated loss of the nigrostriatal pathway and increased sensitivity to MPTP. Neurochem Int 2012, 61, 721-730, doi:10.1016/j.neuint.2012.06.013.
- Scott, T.L.; Wicker, C.A.; Suganya, R.; Dhar, B.; Pittman, T.; Horbinski, C.; Izumi, T. Polyubiquitination of apurinic/apyrimidinic endonuclease 1 by Parkin. Mol Carcinog 2017, 56, 325-336, doi:10.1002/mc.22495.
- Sanders, L.H.; McCoy, J.; Hu, X.; Mastroberardino, P.G.; Dickinson, B.C.; Chang, C.J.; Chu, C.T.; Van Houten, B.; Greenamyre, J.T. Mitochondrial DNA damage: molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiol Dis 2014, 70, 214-223, doi:10.1016/j.nbd.2014.06.014.
- Gencer, M.; Dasdemir, S.; Cakmakoglu, B.; Cetinkaya, Y.; Varlibas, F.; Tireli, H.; Kucukali, C.I.; Ozkok, E.; Aydin, M. DNA repair genes in Parkinson's disease. Genet Test Mol Biomarkers 2012, 16, 504-507, doi:10.1089/gtmb.2011.0252.
- Sanders, L.H.; Paul, K.C.; Howlett, E.H.; Lawal, H.; Boppana, S.; Bronstein, J.M.; Ritz, B.; Greenamyre, J.T. Editor's Highlight: Base Excision Repair Variants and Pesticide Exposure Increase Parkinson's Disease Risk. Toxicol Sci 2017, 158, 188-198, doi:10.1093/toxsci/kfx086.
- Chang, D.; Nalls, M.A.; Hallgrimsdottir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; International Parkinson's Disease Genomics, C.; andMe Research, T.; Kerchner, G.A.; Ayalon, G., et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson's disease risk loci. Nat Genet 2017, 49, 1511-1516, doi:10.1038/ng.3955.
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A., et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson's disease: a meta-analysis of genome-wide association studies. Lancet Neurol 2019, 18, 1091-1102, doi:10.1016/S1474-4422(19)30320-5.
- Vasquez, V.; Mitra, J.; Hegde, P.M.; Pandey, A.; Sengupta, S.; Mitra, S.; Rao, K.S.; Hegde, M.L. Chromatin-Bound Oxidized alpha-Synuclein Causes Strand Breaks in Neuronal Genomes in in vitro Models of Parkinson's Disease. J Alzheimers Dis 2017, 60, S133-S150, doi:10.3233/JAD-170342.
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R., et al. Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson's disease. Science 2018, 362, doi:10.1126/science.aat8407.
- Ragagnin, A.M.G.; Shadfar, S.; Vidal, M.; Jamali, M.S.; Atkin, J.D. Motor Neuron Susceptibility in ALS/FTD. Front Neurosci 2019, 13, 532, doi:10.3389/fnins.2019.00532.
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta 2015, 1852, 679-684, doi:10.1016/j.bbadis.2014.08.010.
- Peters, O.M.; Ghasemi, M.; Brown, R.H., Jr. Emerging mechanisms of molecular pathology in ALS. J Clin Invest 2015, 125, 1767-1779, doi:10.1172/JCI71601.
- van Zundert, B.; Izaurieta, P.; Fritz, E.; Alvarez, F.J. Early pathogenesis in the adult-onset neurodegenerative disease amyotrophic lateral sclerosis. J Cell Biochem 2012, 113, 3301-3312, doi:10.1002/jcb.24234.
- Bradley, W.G.; Krasin, F. A new hypothesis of the etiology of amyotrophic lateral sclerosis. The DNA hypothesis. Arch Neurol 1982, 39, 677-680, doi:10.1001/archneur.1982.00510230003001.
- Kim, B.W.; Jeong, Y.E.; Wong, M.; Martin, L.J. DNA damage accumulates and responses are engaged in human ALS brain and spinal motor neurons and DNA repair is activatable in iPSC-derived motor neurons with SOD1 mutations. Acta Neuropathol Commun 2020, 8, 7, doi:10.1186/s40478-019-0874-4.
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem 1997, 69, 2064-2074, doi:10.1046/j.1471-4159.1997.69052064.x.
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O'Regan, J.P.; Deng, H.X., et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59-62, doi:10.1038/362059a0.
- Brenner, D.; Muller, K.; Wieland, T.; Weydt, P.; Bohm, S.; Lule, D.; Hubers, A.; Neuwirth, C.; Weber, M.; Borck, G., et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain 2016, 139, e28, doi:10.1093/brain/aww033.
- Chen, Y.; Chen, C.F.; Riley, D.J.; Chen, P.L. Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 2011, 10, 655-663, doi:10.4161/cc.10.4.14814.
- Higelin, J.; Catanese, A.; Semelink-Sedlacek, L.L.; Oeztuerk, S.; Lutz, A.K.; Bausinger, J.; Barbi, G.; Speit, G.; Andersen, P.M.; Ludolph, A.C., et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res 2018, 30, 150-162, doi:10.1016/j.scr.2018.06.005.
- Shaikh, A.Y.; Martin, L.J. DNA base-excision repair enzyme apurinic/apyrimidinic endonuclease/redox factor-1 is increased and competent in the brain and spinal cord of individuals with amyotrophic lateral sclerosis. Neuromolecular Med 2002, 2, 47-60, doi:10.1007/s12017-002-0038-7.
- Murakami, T.; Nagai, M.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Kurata, T.; Takehisa, Y.; Kamiya, T.; Abe, K. Early decrease of mitochondrial DNA repair enzymes in spinal motor neurons of presymptomatic transgenic mice carrying a mutant SOD1 gene. Brain Res 2007, 1150, 182-189, doi:10.1016/j.brainres.2007.02.057.
- Mitra, J.; Guerrero, E.N.; Hegde, P.M.; Liachko, N.F.; Wang, H.; Vasquez, V.; Gao, J.; Pandey, A.; Taylor, J.P.; Kraemer, B.C., et al. Motor neuron disease-associated loss of nuclear TDP-43 is linked to DNA double-strand break repair defects. Proc Natl Acad Sci U S A 2019, 116, 4696-4705, doi:10.1073/pnas.1818415116.
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem 2013, 288, 24731-24741, doi:10.1074/jbc.M113.497974.
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gomez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res 2014, 42, 307-314, doi:10.1093/nar/gkt835.
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 2013, 16, 1383-1391, doi:10.1038/nn.3514.
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 function in the DNA-damage response to promote DNA nonhomologous end-joining. Nat Struct Mol Biol 2010, 17, 1144-1151, doi:10.1038/nsmb.1899.
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del'Olio, S.; Huff, T.C.; Chee, N.T., et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol Neurodegener 2020, 15, 13, doi:10.1186/s13024-020-00365-9.
- Farg, M.A.; Konopka, A.; Soo, K.Y.; Ito, D.; Atkin, J.D. The DNA damage response (DDR) is induced by the C9orf72 repeat expansion in amyotrophic lateral sclerosis. Hum Mol Genet 2017, 26, 2882-2896, doi:10.1093/hmg/ddx170.
- Petersen, A.J.; Rimkus, S.A.; Wassarman, D.A. ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc Natl Acad Sci U S A 2012, 109, E656-664, doi:10.1073/pnas.1110470109.
- Illuzzi, J.; Yerkes, S.; Parekh-Olmedo, H.; Kmiec, E.B. DNA breakage and induction of DNA damage response proteins precede the appearance of visible mutant huntingtin aggregates. J Neurosci Res 2009, 87, 733-747, doi:10.1002/jnr.21881.
- Lu, X.H.; Mattis, V.B.; Wang, N.; Al-Ramahi, I.; van den Berg, N.; Fratantoni, S.A.; Waldvogel, H.; Greiner, E.; Osmand, A.; Elzein, K., et al. Targeting ATM ameliorates mutant Huntingtin toxicity in cell and animal models of Huntington's disease. Sci Transl Med 2014, 6, 268ra178, doi:10.1126/scitranslmed.3010523.
- Milanese, C.; Cerri, S.; Ulusoy, A.; Gornati, S.V.; Plat, A.; Gabriels, S.; Blandini, F.; Di Monte, D.A.; Hoeijmakers, J.H.; Mastroberardino, P.G. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson's disease. Cell Death Dis 2018, 9, 818, doi:10.1038/s41419-018-0848-7.
- Mano, T.; Nagata, K.; Nonaka, T.; Tarutani, A.; Imamura, T.; Hashimoto, T.; Bannai, T.; Koshi-Mano, K.; Tsuchida, T.; Ohtomo, R., et al. Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer's disease. Proc Natl Acad Sci U S A 2017, 114, E9645-E9654, doi:10.1073/pnas.1707151114.
- Mao, P.; Reddy, P.H. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer's disease: implications for early intervention and therapeutics. Biochim Biophys Acta 2011, 1812, 1359-1370, doi:10.1016/j.bbadis.2011.08.005.
- Kurihara, M.; Mano, T.; Saito, Y.; Murayama, S.; Toda, T.; Iwata, A. Colocalization of BRCA1 with Tau Aggregates in Human Tauopathies. Brain Sci 2019, 10, doi:10.3390/brainsci10010007.
- Nakamura, M.; Kaneko, S.; Dickson, D.W.; Kusaka, H. Aberrant Accumulation of BRCA1 in Alzheimer Disease and Other Tauopathies. J Neuropathol Exp Neurol 2020, 79, 22-33, doi:10.1093/jnen/nlz107.
- Wezyk, M.; Szybinska, A.; Wojsiat, J.; Szczerba, M.; Day, K.; Ronnholm, H.; Kele, M.; Berdynski, M.; Peplonska, B.; Fichna, J.P., et al. Overactive BRCA1 Affects Presenilin 1 in Induced Pluripotent Stem Cell-Derived Neurons in Alzheimer's Disease. J Alzheimers Dis 2018, 62, 175-202, doi:10.3233/JAD-170830.
- Noristani, H.N.; Sabourin, J.C.; Gerber, Y.N.; Teigell, M.; Sommacal, A.; Vivanco, M.; Weber, M.; Perrin, F.E. Brca1 is expressed in human microglia and is dysregulated in human and animal model of ALS. Mol Neurodegener 2015, 10, 34, doi:10.1186/s13024-015-0023-x.
- Brooks, P.J. DNA repair in neural cells: basic science and clinical implications. Mutat Res 2002, 509, 93-108, doi:10.1016/s0027-5107(02)00222-1.
- Shackelford, D.A. DNA end joining activity is reduced in Alzheimer's disease. Neurobiol Aging 2006, 27, 596-605, doi:10.1016/j.neurobiolaging.2005.03.009.
- Davydov, V.; Hansen, L.A.; Shackelford, D.A. Is DNA repair compromised in Alzheimer's disease? Neurobiol Aging 2003, 24, 953-968, doi:10.1016/s0197-4580(02)00229-4.
- Cardinale, A.; Racaniello, M.; Saladini, S.; De Chiara, G.; Mollinari, C.; de Stefano, M.C.; Pocchiari, M.; Garaci, E.; Merlo, D. Sublethal doses of beta-amyloid peptide abrogate DNA-dependent protein kinase activity. J Biol Chem 2012, 287, 2618-2631, doi:10.1074/jbc.M111.276550.
- Altmeyer, M.; Messner, S.; Hassa, P.O.; Fey, M.; Hottiger, M.O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res 2009, 37, 3723-3738, doi:10.1093/nar/gkp229.
- Aredia, F.; Scovassi, A.I. Involvement of PARPs in cell death. Front Biosci (Elite Ed) 2014, 6, 308-317, doi:10.2741/707.
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci 2008, 13, 3046-3082, doi:10.2741/2909.
- Harrision, D.; Gravells, P.; Thompson, R.; Bryant, H.E. Poly(ADP-Ribose) Glycohydrolase (PARG) vs. Poly(ADP-Ribose) Polymerase (PARP) - Function in Genome Maintenance and Relevance of Inhibitors for Anti-cancer Therapy. Front Mol Biosci 2020, 7, 191, doi:10.3389/fmolb.2020.00191.
- Beneke, S. Regulation of chromatin structure by poly(ADP-ribosyl)ation. Front Genet 2012, 3, 169, doi:10.3389/fgene.2012.00169.
- Burkle, A.; Virag, L. Poly(ADP-ribose): PARadigms and PARadoxes. Mol Aspects Med 2013, 34, 1046-1065, doi:10.1016/j.mam.2012.12.010.
- Fang, E.F.; Kassahun, H.; Croteau, D.L.; Scheibye-Knudsen, M.; Marosi, K.; Lu, H.; Shamanna, R.A.; Kalyanasundaram, S.; Bollineni, R.C.; Wilson, M.A., et al. NAD(+) Replenishment Improves Lifespan and Healthspan in Ataxia Telangiectasia Models via Mitophagy and DNA Repair. Cell Metab 2016, 24, 566-581, doi:10.1016/j.cmet.2016.09.004.
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci 2010, 30, 2967-2978, doi:10.1523/JNEUROSCI.5552-09.2010.
- Croteau, D.L.; Fang, E.F.; Nilsen, H.; Bohr, V.A. NAD(+) in DNA repair and mitochondrial maintenance. Cell Cycle 2017, 16, 491-492, doi:10.1080/15384101.2017.1285631.
- Love, S.; Barber, R.; Wilcock, G.K. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain 1999, 122 ( Pt 2), 247-253, doi:10.1093/brain/122.2.247.
- Kauppinen, T.M.; Suh, S.W.; Higashi, Y.; Berman, A.E.; Escartin, C.; Won, S.J.; Wang, C.; Cho, S.H.; Gan, L.; Swanson, R.A. Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid beta. J Neuroinflammation 2011, 8, 152, doi:10.1186/1742-2094-8-152.
- Martire, S.; Fuso, A.; Rotili, D.; Tempera, I.; Giordano, C.; De Zottis, I.; Muzi, A.; Vernole, P.; Graziani, G.; Lococo, E., et al. PARP-1 modulates amyloid beta peptide-induced neuronal damage. PLoS One 2013, 8, e72169, doi:10.1371/journal.pone.0072169.
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X., et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat Neurosci 2019, 22, 401-412, doi:10.1038/s41593-018-0332-9.
- Mandir, A.S.; Przedborski, S.; Jackson-Lewis, V.; Wang, Z.Q.; Simbulan-Rosenthal, C.M.; Smulson, M.E.; Hoffman, B.E.; Guastella, D.B.; Dawson, V.L.; Dawson, T.M. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A 1999, 96, 5774-5779, doi:10.1073/pnas.96.10.5774.
- Shibata, N.; Kakita, A.; Takahashi, H.; Ihara, Y.; Nobukuni, K.; Fujimura, H.; Sakoda, S.; Sasaki, S.; Yamamoto, T.; Kobayashi, M. Persistent cleavage and nuclear translocation of apoptosis-inducing factor in motor neurons in the spinal cord of sporadic amyotrophic lateral sclerosis patients. Acta Neuropathol 2009, 118, 755-762, doi:10.1007/s00401-009-0580-6.
- McGurk, L.; Mojsilovic-Petrovic, J.; Van Deerlin, V.M.; Shorter, J.; Kalb, R.G.; Lee, V.M.; Trojanowski, J.Q.; Lee, E.B.; Bonini, N.M. Nuclear poly(ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol Commun 2018, 6, 84, doi:10.1186/s40478-018-0586-1.
- Oh, Y.K.; Shin, K.S.; Kang, S.J. AIF translocates to the nucleus in the spinal motor neurons in a mouse model of ALS. Neurosci Lett 2006, 406, 205-210, doi:10.1016/j.neulet.2006.07.044.
- Schondorf, D.C.; Ivanyuk, D.; Baden, P.; Sanchez-Martinez, A.; De Cicco, S.; Yu, C.; Giunta, I.; Schwarz, L.K.; Di Napoli, G.; Panagiotakopoulou, V., et al. The NAD+ Precursor Nicotinamide Riboside Rescues Mitochondrial Defects and Neuronal Loss in iPSC and Fly Models of Parkinson's Disease. Cell Rep 2018, 23, 2976-2988, doi:10.1016/j.celrep.2018.05.009.
- Menzies, F.M.; Fleming, A.; Rubinsztein, D.C. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 2015, 16, 345-357, doi:10.1038/nrn3961.
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B., et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 2008, 118, 2190-2199, doi:10.1172/JCI33585.
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat Med 2013, 19, 983-997, doi:10.1038/nm.3232.
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 2008, 283, 23542-23556, doi:10.1074/jbc.M801992200.
- Fifita, J.A.; Williams, K.L.; Sundaramoorthy, V.; McCann, E.P.; Nicholson, G.A.; Atkin, J.D.; Blair, I.P. A novel amyotrophic lateral sclerosis mutation in OPTN induces ER stress and Golgi fragmentation in vitro. Amyotroph Lateral Scler Frontotemporal Degener 2017, 18, 126-133, doi:10.1080/21678421.2016.1218517.
- Korac, J.; Schaeffer, V.; Kovacevic, I.; Clement, A.M.; Jungblut, B.; Behl, C.; Terzic, J.; Dikic, I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci 2013, 126, 580-592, doi:10.1242/jcs.114926.
- Goode, A.; Butler, K.; Long, J.; Cavey, J.; Scott, D.; Shaw, B.; Sollenberger, J.; Gell, C.; Johansen, T.; Oldham, N.J., et al. Defective recognition of LC3B by mutant SQSTM1/p62 implicates impairment of autophagy as a pathogenic mechanism in ALS-FTLD. Autophagy 2016, 12, 1094-1104, doi:10.1080/15548627.2016.1170257.
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S., et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74-79, doi:10.1038/nature09803.
- Orlotti, N.I.; Cimino-Reale, G.; Borghini, E.; Pennati, M.; Sissi, C.; Perrone, F.; Palumbo, M.; Daidone, M.G.; Folini, M.; Zaffaroni, N. Autophagy acts as a safeguard mechanism against G-quadruplex ligand-mediated DNA damage. Autophagy 2012, 8, 1185-1196, doi:10.4161/auto.20519.
- Eapen, V.V.; Haber, J.E. DNA damage signaling triggers the cytoplasm-to-vacuole pathway of autophagy to regulate cell cycle progression. Autophagy 2013, 9, 440-441, doi:10.4161/auto.23280.
- Alexander, A.; Cai, S.L.; Kim, J.; Nanez, A.; Sahin, M.; MacLean, K.H.; Inoki, K.; Guan, K.L.; Shen, J.; Person, M.D., et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A 2010, 107, 4153-4158, doi:10.1073/pnas.0913860107.
- Alexander, A.; Kim, J.; Walker, C.L. ATM engages the TSC2/mTORC1 signaling node to regulate autophagy. Autophagy 2010, 6, 672-673, doi:10.4161/auto.6.5.12509.
- Cheng, A.; Tse, K.H.; Chow, H.M.; Gan, Y.; Song, X.; Ma, F.; Qian, Y.X.Y.; She, W.; Herrup, K. ATM loss disrupts the autophagy-lysosomal pathway. Autophagy 2020, 1-13, doi:10.1080/15548627.2020.1805860.
- Rodriguez-Vargas, J.M.; Ruiz-Magana, M.J.; Ruiz-Ruiz, C.; Majuelos-Melguizo, J.; Peralta-Leal, A.; Rodriguez, M.I.; Munoz-Gamez, J.A.; de Almodovar, M.R.; Siles, E.; Rivas, A.L., et al. ROS-induced DNA damage and PARP-1 are required for optimal induction of starvation-induced autophagy. Cell Res 2012, 22, 1181-1198, doi:10.1038/cr.2012.70.
- Xu, F.; Fang, Y.; Yan, L.; Xu, L.; Zhang, S.; Cao, Y.; Xu, L.; Zhang, X.; Xie, J.; Jiang, G., et al. Nuclear localization of Beclin 1 promotes radiation-induced DNA damage repair independent of autophagy. Sci Rep 2017, 7, 45385, doi:10.1038/srep45385.
- SenGupta, T.; Torgersen, M.L.; Kassahun, H.; Vellai, T.; Simonsen, A.; Nilsen, H. Base excision repair AP endonucleases and mismatch repair act together to induce checkpoint-mediated autophagy. Nat Commun 2013, 4, 2674, doi:10.1038/ncomms3674.
- Hewitt, G.; Carroll, B.; Sarallah, R.; Correia-Melo, C.; Ogrodnik, M.; Nelson, G.; Otten, E.G.; Manni, D.; Antrobus, R.; Morgan, B.A., et al. SQSTM1/p62 mediates crosstalk between autophagy and the UPS in DNA repair. Autophagy 2016, 12, 1917-1930, doi:10.1080/15548627.2016.1210368.
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun 2015, 6, 6823, doi:10.1038/ncomms7823.
- Zhao, Z.; Ni, D.; Ghozalli, I.; Pirooz, S.D.; Ma, B.; Liang, C. UVRAG: at the crossroad of autophagy and genomic stability. Autophagy 2012, 8, 1392-1393, doi:10.4161/auto.21035.
- Dan, X.; Babbar, M.; Moore, A.; Wechter, N.; Tian, J.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. DNA damage invokes mitophagy through a pathway involving Spata18. Nucleic Acids Res 2020, 48, 6611-6623, doi:10.1093/nar/gkaa393.
- Chaurasia, M.; Gupta, S.; Das, A.; Dwarakanath, B.S.; Simonsen, A.; Sharma, K. Radiation induces EIF2AK3/PERK and ERN1/IRE1 mediated pro-survival autophagy. Autophagy 2019, 15, 1391-1406, doi:10.1080/15548627.2019.1582973.
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 2004, 3, 205-214, doi:10.1038/nrd1330.
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a Common Feature of Neurodegenerative Disorders. Front Pharmacol 2019, 10, 1008, doi:10.3389/fphar.2019.01008.
- Mikhed, Y.; Gorlach, A.; Knaus, U.G.; Daiber, A. Redox regulation of genome stability by effects on gene expression, epigenetic pathways and DNA damage/repair. Redox Biol 2015, 5, 275-289, doi:10.1016/j.redox.2015.05.008.
- Moritz, E.; Pauly, K.; Bravard, A.; Hall, J.; Radicella, J.P.; Epe, B. hOGG1-Cys326 variant cells are hypersensitive to DNA repair inhibition by nitric oxide. Carcinogenesis 2014, 35, 1426-1433, doi:10.1093/carcin/bgu066.
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786-791, doi:10.1126/science.1232458.
- Chin, A.C. Neuroinflammation and the cGAS-STING pathway. J Neurophysiol 2019, 121, 1087-1091, doi:10.1152/jn.00848.2018.
- Quek, H.; Luff, J.; Cheung, K.; Kozlov, S.; Gatei, M.; Lee, C.S.; Bellingham, M.C.; Noakes, P.G.; Lim, Y.C.; Barnett, N.L., et al. A rat model of ataxia-telangiectasia: evidence for a neurodegenerative phenotype. Hum Mol Genet 2017, 26, 109-123, doi:10.1093/hmg/ddw371.
- Xu, Q.; Xu, W.; Cheng, H.; Yuan, H.; Tan, X. Efficacy and mechanism of cGAMP to suppress Alzheimer's disease by elevating TREM2. Brain Behav Immun 2019, 81, 495-508, doi:10.1016/j.bbi.2019.07.004.
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D., et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636-649 e618, doi:10.1016/j.cell.2020.09.020.
- Borsche, M.; Konig, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Bruggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M., et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020, 143, 3041-3051, doi:10.1093/brain/awaa246.
- Liachko, N.F.; Guthrie, C.R.; Kraemer, B.C. Phosphorylation promotes neurotoxicity in a Caenorhabditis elegans model of TDP-43 proteinopathy. J Neurosci 2010, 30, 16208-16219, doi:10.1523/JNEUROSCI.2911-10.2010.
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell 2014, 157, 882-896, doi:10.1016/j.cell.2014.03.026.
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann Neurol 2013, 74, 180-187, doi:10.1002/ana.23946.
- Imamura, S.; Kishi, S. Molecular cloning and functional characterization of zebrafish ATM. Int J Biochem Cell Biol 2005, 37, 1105-1116, doi:10.1016/j.biocel.2004.10.015.
- Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirvio, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem 2003, 86, 165-172, doi:10.1046/j.1471-4159.2003.01809.x.
- van Ham, T.J.; Thijssen, K.L.; Breitling, R.; Hofstra, R.M.; Plasterk, R.H.; Nollen, E.A. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet 2008, 4, e1000027, doi:10.1371/journal.pgen.1000027.
- McColl, G.; Roberts, B.R.; Pukala, T.L.; Kenche, V.B.; Roberts, C.M.; Link, C.D.; Ryan, T.M.; Masters, C.L.; Barnham, K.J.; Bush, A.I., et al. Utility of an improved model of amyloid-beta (Abeta(1)(-)(4)(2)) toxicity in Caenorhabditis elegans for drug screening for Alzheimer's disease. Mol Neurodegener 2012, 7, 57, doi:10.1186/1750-1326-7-57.
- Wu, Y.; Wu, Z.; Butko, P.; Christen, Y.; Lambert, M.P.; Klein, W.L.; Link, C.D.; Luo, Y. Amyloid-beta-induced pathological behaviors are suppressed by Ginkgo biloba extract EGb 761 and ginkgolides in transgenic Caenorhabditis elegans. J Neurosci 2006, 26, 13102-13113, doi:10.1523/JNEUROSCI.3448-06.2006.
- Shen, L.; Wang, C.; Chen, L.; Leung, K.L.; Lo, E.; Lakso, M.; Wong, G. TDP-1/TDP-43 potentiates human alpha-Synuclein (HASN) neurodegeneration in Caenorhabditis elegans. Biochim Biophys Acta Mol Basis Dis 2020, 1866, 165876, doi:10.1016/j.bbadis.2020.165876.
- Muqit, M.M.; Feany, M.B. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat Rev Neurosci 2002, 3, 237-243, doi:10.1038/nrn751.
- Pitchai, A.; Rajaretinam, R.K.; Freeman, J.L. Zebrafish as an Emerging Model for Bioassay-Guided Natural Product Drug Discovery for Neurological Disorders. Medicines (Basel) 2019, 6, doi:10.3390/medicines6020061.
- Saleem, S.; Kannan, R.R. Zebrafish: an emerging real-time model system to study Alzheimer's disease and neurospecific drug discovery. Cell Death Discov 2018, 4, 45, doi:10.1038/s41420-018-0109-7.
- Xi, Y.; Noble, S.; Ekker, M. Modeling neurodegeneration in zebrafish. Curr Neurol Neurosci Rep 2011, 11, 274-282, doi:10.1007/s11910-011-0182-2.
- Abugable, A.A.; Morris, J.L.M.; Palminha, N.M.; Zaksauskaite, R.; Ray, S.; El-Khamisy, S.F. DNA repair and neurological disease: From molecular understanding to the development of diagnostics and model organisms. DNA Repair (Amst) 2019, 81, 102669, doi:10.1016/j.dnarep.2019.102669.
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; Van Gorp, S.; Nazor, K.L.; Boscolo, F.S., et al. Probing sporadic and familial Alzheimer's disease using induced pluripotent stem cells. Nature 2012, 482, 216-220, doi:10.1038/nature10821.
- Kimura, J.; Shimizu, K.; Kajima, K.; Yokosuka, A.; Mimaki, Y.; Oku, N.; Ohizumi, Y. Nobiletin Reduces Intracellular and Extracellular beta-Amyloid in iPS Cell-Derived Alzheimer's Disease Model Neurons. Biol Pharm Bull 2018, 41, 451-457, doi:10.1248/bpb.b17-00364.
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Reports 2017, 8, 856-869, doi:10.1016/j.stemcr.2017.02.019.
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Gunther, R.; Jin, M.; Stanslowsky, N., et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat Commun 2018, 9, 335, doi:10.1038/s41467-017-02299-1.
- Seminary, E.R.; Sison, S.L.; Ebert, A.D. Modeling Protein Aggregation and the Heat Shock Response in ALS iPSC-Derived Motor Neurons. Front Neurosci 2018, 12, 86, doi:10.3389/fnins.2018.00086.
- Doi, D.; Magotani, H.; Kikuchi, T.; Ikeda, M.; Hiramatsu, S.; Yoshida, K.; Amano, N.; Nomura, M.; Umekage, M.; Morizane, A., et al. Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson's disease. Nat Commun 2020, 11, 3369, doi:10.1038/s41467-020-17165-w.
- Lopez de Maturana, R.; Lang, V.; Zubiarrain, A.; Sousa, A.; Vazquez, N.; Gorostidi, A.; Aguila, J.; Lopez de Munain, A.; Rodriguez, M.; Sanchez-Pernaute, R. Mutations in LRRK2 impair NF-kappaB pathway in iPSC-derived neurons. J Neuroinflammation 2016, 13, 295, doi:10.1186/s12974-016-0761-x.
- Lin, Y.T.; Seo, J.; Gao, F.; Feldman, H.M.; Wen, H.L.; Penney, J.; Cam, H.P.; Gjoneska, E.; Raja, W.K.; Cheng, J., et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer's Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 2018, 98, 1294, doi:10.1016/j.neuron.2018.06.011.
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol Med 2017, 23, 393-410, doi:10.1016/j.molmed.2017.02.007.
- Ormel, P.R.; Vieira de Sa, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A., et al. Microglia innately develop within cerebral organoids. Nat Commun 2018, 9, 4167, doi:10.1038/s41467-018-06684-2.
- Yan, Y.; Song, L.; Bejoy, J.; Zhao, J.; Kanekiyo, T.; Bu, G.; Zhou, Y.; Li, Y. Modeling Neurodegenerative Microenvironment Using Cortical Organoids Derived from Human Stem Cells. Tissue Eng Part A 2018, 24, 1125-1137, doi:10.1089/ten.TEA.2017.0423.
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol Psychiatry 2018, 23, 2363-2374, doi:10.1038/s41380-018-0229-8.
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer's disease with iPSC-derived brain cells. Mol Psychiatry 2020, 25, 148-167, doi:10.1038/s41380-019-0468-3.
- Kim, H.; Park, H.J.; Choi, H.; Chang, Y.; Park, H.; Shin, J.; Kim, J.; Lengner, C.J.; Lee, Y.K.; Kim, J. Modeling G2019S-LRRK2 Sporadic Parkinson's Disease in 3D Midbrain Organoids. Stem Cell Reports 2019, 12, 518-531, doi:10.1016/j.stemcr.2019.01.020.
- Monzel, A.S.; Smits, L.M.; Hemmer, K.; Hachi, S.; Moreno, E.L.; van Wuellen, T.; Jarazo, J.; Walter, J.; Bruggemann, I.; Boussaad, I., et al. Derivation of Human Midbrain-Specific Organoids from Neuroepithelial Stem Cells. Stem Cell Reports 2017, 8, 1144-1154, doi:10.1016/j.stemcr.2017.03.010.
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J., et al. Modeling Parkinson's disease in midbrain-like organoids. NPJ Parkinsons Dis 2019, 5, 5, doi:10.1038/s41531-019-0078-4.
- Osaki, T.; Sivathanu, V.; Kamm, R.D. Engineered 3D vascular and neuronal networks in a microfluidic platform. Sci Rep 2018, 8, 5168, doi:10.1038/s41598-018-23512-1.
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.; Fisher, E.M.; Strydom, A. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci 2015, 16, 564-574, doi:10.1038/nrn3983.
- Bandmann, O.; Davis, M.B.; Marsden, C.D.; Harding, A.E. Sequence of the superoxide dismutase 1 (SOD 1) gene in familial Parkinson's disease. J Neurol Neurosurg Psychiatry 1995, 59, 90-91, doi:10.1136/jnnp.59.1.90.
- Choi, J.; Rees, H.D.; Weintraub, S.T.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative modifications and aggregation of Cu,Zn-superoxide dismutase associated with Alzheimer and Parkinson diseases. J Biol Chem 2005, 280, 11648-11655, doi:10.1074/jbc.M414327200.