Cellular senescence, one of the hallmarks of aging, is defined as a cellular state of irreversibly arrested proliferation of aged or damaged cells.
- cellular senescence
- lung fibrosis
- pathogenesis
1. Introduction
Cellular senescence, one of the hallmarks of aging [1], is defined as a cellular state of irreversibly arrested proliferation of aged or damaged cells [2]. Senescent cells have distinct phenotypic alterations, including genomic instability, telomere attrition, chromatin remodeling, metabolic reprogramming, increased autophagy, decreased mitophagy, and the implementation of a complex pro-inflammatory secretome [3][4][10,11]. Although senescence was first characterized as a state of cell cycle arrest after extensive proliferation, referred to as replicative senescence (RS) , this cellular process can be induced by different stimuli, including telomere attrition, DNA damage caused by strong genotoxic stress, such as ionizing radiation/IRIS, topoisomerase inhibitors, and oxidative agents/OSIS [5][13], chromatin perturbations, and oncogene activation/OIS.
Interstitial lung diseases (ILDs) and pulmonary fibrosis are heterogeneous groups of lung diseases characterized by varying degrees of inflammation and fibrosis of the lung parenchyma. Some of these may occur secondary to a known precipitant such as medications, autoimmune or connective tissue disease, hypersensitivity to inhaled organic antigens, or sarcoidosis, while others have no identifiable cause and are classified as idiopathic interstitial pneumonias (IIPs) [6][14]. Idiopathic pulmonary fibrosis (IPF) is one of the most aggressive forms of IIP, with a median survival time of 2–3 years after diagnosis [6][14]. Features of this pulmonary disease include temporal and spatially heterogeneous fibrosis, the presence of fibroblastic foci, and the excessive deposition of disorganized collagen and extracellular matrix (ECM), which result in the loss of normal lung architecture, with or without honeycomb cyst formation [7][15].
Multiple risk factors have been reported to increase the risk of the development of IPF in an independent or coordinated fashion. The endogenous risk factors include genetic background, aging, gender, and pulmonary microbiology, whereas the exogenous factors include cigarette smoking, environmental exposure, and air pollution, especially the dust or organic solvents exposure in the occupational population. Comorbidities such as obstructive sleep apnea, gastroesophageal reflux, and diabetes mellitus are also important factors [8][16]. A longitudinal study to identify independent risk factors of ILD development found that people over 70 had a 6.9 times higher risk for ILD than those over 40 years old [9][17].
It has been long recognized that cellular senescence contributes to an aging-related decline in tissue regeneration capacity and the pathogenesis of aging-related diseases, including IPF (reviewed in [10][21]). However, the mechanism whereby senescent cells contribute individually or in a coordinated fashion or both to lung fibrosis remains unclear. Herein, we review the current knowledge on the senescence of the primary cell types in the lung, their phenotypic changes, and the cellular and molecular mechanisms by which these cells contribute to the pathogenesis of pulmonary fibrosis.
2. Senescence Regulatory Pathways
The complexity of cellular senescence regulation is well-characterized in many disease models, especially aging [11][12][22,23]. As mentioned earlier, multiple internal (genetic abnormalities) and external stressors (oncogene activation, oxidative stress, etc.) are the commonly known trigger factors of cellular senescence (Figure 1). Our review discusses only the studied regulatory pathways in each prominent cell type significantly involved in lung fibrosis pathogenesis. We find that each cell type uses at least one of both pathways, but nearly all lung cells use the SASP, suggesting the intimate interconnection of these cells in regulating lung fibrosis through cellular senescence.
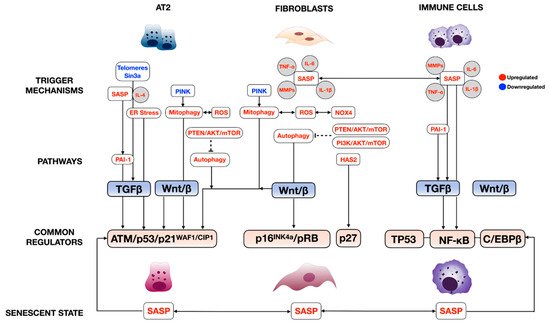
The regulation of senescence through the cell cycle is mediated through classical cell signaling molecules such as DNA-damage response (DDR), These signaling molecules directly activate cell cycle inhibitors, including p14, p15, p16, p17, p21, and p27, and indirectly through TP53 [11][12][22,23]. Phosphorylated Rb loses its ability to associate with a cell pro-proliferative transcriptional factor, E2F1-3, thereby averting normal cell cycle progression (Figure 1) This pathway is well-described in crucial cellular players in lung fibrosis, including alveolar type 2 cells (AT2), lung fibroblasts, and immune cells; further details are discussed inSection 4, “
The SASP has been described in multiple senescent cell types in the lung. The prominent SASPs in fibroproliferative lung diseases, which include growth factors such as TGFβ and many inflammatory cytokines/chemokines such as IL-6, TNF-α, and MMPs, mainly regulate senescence through TP53 and NF-κB In this case, IL-4 and IL-13, released from senescent AT2, promotes alveolar macrophage activation in lung fibrosis via the PAI-1/TGFβ1 axis [13][38]. C/EBPβ also plays an important role in experimental lung fibrosis, as demonstrated by C/EBPβ null mice that developed less lung fibrosis after bleomycin injury due to the reduced production of lung pro-inflammatory cytokines TGFβ1, TNF-α, and IL-1β, and myofibroblast differentiation [14][39].
3. The Role of Cellular Senescence in the Pathogenesis of Pulmonary Fibrosis
Recent studies have indicated that the initiation and/or progression of IPF are linked to epithelial progenitor cell dysfunction. Accelerating aging, including the loss of epithelial progenitor cell function and/or numbers and cellular senescence, is one of the key factors in IPF pathogenesis [15][16][17][40,41,42]. Furthermore, multiple genome-wide screens for IPF-associated gene variants have defined a serial of gene variants linked to defects in the host defense and regulation of cellular senescence, as well as epithelial cell function [18][19][43,44]. In up to 15–20% of the familial IPF cases, causative mutations have been identified in genes involved in regulating telomere function—telomerase reverse transcriptase, TERT; telomerase RNA component, TERC; dyskerin, DKC1; telomere interacting factor 2, TINF2; and regulator of telomere elongation helicase, RTEL1.
The AT2 cell, a facultative stem cell that produces and secretes pulmonary surfactants in its quiescent state, is considered the progenitor cell responsible for normal maintenance of the alveolar epithelium [20][48]. Genetic or environmental factors can compromise its ability to proliferate and can lead to defective alveolar epithelium maintenance. Recent studies using single-cell RNAseq have shown both the regional depletion of AT2 cells and abnormal activation of cellular senescence and senescence-related signaling pathways such as p53 signaling, mitochondria dysfunction, and oxidative stress, in the fibrotic region of IPF explant tissue [21][22][23][27,49,50], also evidenced by increased SA-β-gal signaling and the expression of p16 and p21 in SFTPC-positive AT2 cells. Different genetic in vivo animal models (Table 1) and in vitro precision-cut lung slides models have been developed to demonstrate AT2 cell dysfunction, especially cellular senescence in the initiation and progression of pulmonary fibrosis.
| Animal Models | Spontaneous Fibrosis | Induced Fibrosis | References |
|---|---|---|---|
| Tert−/− | No | 4th-generation inbred, reduced required dose of bleomycin to induce fibrosis | [24] |
| Terc−/− | No | 3rd-generation enhanced fibrosis upon liposaccharide and bleomycin | [25] |
| SftpcCerER; Tert flox/flox | No | Bleomycin induced enhanced fibrosis | [26] |
| SftpcCerER; Trf1flox/flox | Yes | [24][27] | |
| SftpcCerER; Trf2flox/flox | No | Increased susceptibility to bleomycin | [28] |
| SftpcCerER; Grp78flox/flox | Yes | [29] | |
| SftpcCerER; Sin3aflox/flox | Yes | [30] | |
| mSFTPC.rtTA; SFTPCL188Q |
No | Bleomycin induced exaggerated lung fibrosis | [31] |
| SftpcCerER; SFTPCI73T | Yes | [32] | |
| SftpcCerER; SFTPCC121G | Yes | [33] |
The shortening of telomeres has been tightly linked to the induction of cellular senescence [34][59]. Animal models focusing on the role of telomere function have also demonstrated the causal relationship of telomere disfunction in AT2 cells for pulmonary fibrosis. ) mice develop spontaneous lung fibrosis [30][26][61,62], even with inbred mice up to the fourth to the sixth generation of Tert−/−with the requirement of bleomycin dosage to induce lung fibrosis [35][51]. The loss of Trf1 in AT2 cells leads to spontaneous pulmonary fibrosis with the activation of DNA damage, indicated by the increased expression of γH2ax, short telomeres, and accumulation of senescent cells [35][36][51,64].
The perturbation of ER homeostasis by stimuli such as mutations, hypoxia, oxidative stress, chaperone shortage, and unfolded or misfolded proteins leads to a stress condition called “ER stress”, which is toxic and detrimental to cells. The unfolded protein response (UPR) is one of the programs that aim to restore ER’s physiological activity—protein homeostasis (proteostasis). In a transgenic mouse model, the induced expression of mutant SFTPCL188Qin AT2 cells induces ER stress and increases bleomycin susceptibility [37][68]; the induced expression of mutant SFTPC variants SFTPCI73Tand SFTPCC121Gin adult AT2 cells leads to spontaneous pulmonary fibrosis with the activation of ER stress and induction of AT2 cells secretion of SASP-related cytokines [38][39][56,57]. The deletion of Grp78, the chaperon protein of UPR, from AT2 cells leads to spontaneous lung fibrosis.
The excessive production of reactive oxygen species (ROS), which is called oxidative stress, is another critical inducer of cellular senescence [40][55]. Chronic ROS exposure can lead to oxidative damage to the mitochondria due to the sensitivity of mitochondrial membrane phospholipids to reactive species and can further induce more ROS production [41][69]. The ectopic activation of oxidative stress and the mitochondrial dysfunction signal pathway has been observed in IPF AT2 cells compared to donor AT2 cells [21][27]. The loss of Pink1 in mouse AT2 cells results in mitochondrial dysfunction, upregulation of senescence markers (p16 and p21), and increased levels of TGF-β expression [42][43][71,72].
The acetylation of p53 has been shown to play an essential role in the stabilization and activation of p53 in fibrotic lungs [21][23][44][45][27,50,73,74]. A decreased SIRT1 expression level has been observed in IPF [44][73], and the activation of Sirt1 expression by Sirt1 activator reduces lung fibrosis in the bleomycin-induced lung injury mouse model [46][47][76,77]. The loss of Sin3a results in an increased acetylation level of p53 [48][79], whereas the simultaneous knockout of Sin3a and p53 in AT2 cells reduces lung fibrosis. These data demonstrate that the acetylation of p53 in AT2 cells may play an important role in the activation of the p53 pathway and in driving AT2 cell senescence in pulmonary fibrosis.
Airway basal and basal-like cells are gaining interest in lung fibrosis pathogenesis as these cells are aberrantly expanding in IPF. The abnormally increased proliferation of airway basal cells such as classic murine Trp63+Krt5+and human p63+KRT5+cells, human KRT5+KRT14+p63+[23][49][50,81], KRT15+[50][82], and KRT17+[50][51][52][82,83,84] have emerged in lung repairing processes. Although the proliferation of these basal cells during lung fibrosis argues against the senescent characteristics, two studies indicated that these KRT15+and KRT17+basal cells expressed cellular senescence characteristics at the transcriptomic [50][82] and protein level [51][83], suggesting their potential roles in IPF pathogenesis.
More recently, one single-cell RNA-seq cohort identified aberrant basaloid cells in IPF (VIM, CDH2, FN1, COL1A1, TNC, and HMGA2), and senescence (CDKN1A, CDKN2A, CCND1, CCND2, MDM2, and GDF15) Another study also described aberrant basaloid cells in IPF and systemic sclerosis-related ILDs, whereby those cells expressed senescence markers, CDKN2A (gene for p16), CDKN1A (gene for p21), andGDF15[53][86], and that cellular senescence was one of the profibrotic regulatory pathways identified in these cells. Although pathological functions of these basal cell subtypes remain to be explored, accumulating evidence supports their involvement in lung fibrosis through the cellular senescence pathway.
Fibroblasts have a pivotal role in wound healing in response to lung injury. At the site, fibroblasts differentiate into myofibroblasts, which exhibit contractile function and produce ECM components that are essential to wound healing [54][88]. As the normal repair process progresses, myofibroblasts gradually become senescent, which reduces fibroblast activation, ECM deposition, and limits the progression of fibrosis. However, studies showing that targeting senescent fibroblasts and/or myofibroblasts with senolytic drugs significantly ameliorates pulmonary fibrosis and function in mice models of IPF have revealed a deleterious role for these cells in lung fibrosis [16][55][41,92].
Numerous studies have characterized fibroblasts derived from fibrotic lung diseases such as IPF and compared them to age-matched controls. Overall, there is a consensus that fibroblast senescence is increased [56][57][93,94] and persistent in the IPF lung [16][56][41,93]. In vitro, these cells exhibit an enlarged flattened morphology, lower cell division rates, and increased expression of SA-β-galactosidase compared to control fibroblasts at the same passage [55][57][58][92,94,95]. In addition to the aforementioned hallmark features of cellular senescence, fibroblasts from IPF lungs also exhibit metabolic reprogramming, mitochondrial dysfunction, insufficient autophagy, and reduced apoptosis [55][57][92,94].
Although the SASP plays an important physiological role, such as cell proliferation and differentiation during wound repair, its persistence in the environment may contribute to IPF pathology. Senescent IPF fibroblasts secrete numerous pro-inflammatory cytokines/chemokines, pro-fibrotic factors, and reactive oxygen species [16][55][57][58][41,92,94,95]. It has been reported that the SASP can spread senescence signals to surrounding cells, contribute to persistent inflammation [24][96] and tissue remodeling [58][95], and promote profibrotic phenotypic changes of fibroblasts and/or macrophages. Importantly, the deficiency in cytotoxic NK activity could be one of the altered mechanisms perpetrating the accumulation of senescent fibroblasts and other cell types in IPF lungs.
Mitochondria are a central hub not only for cellular energy but also for multiple signaling pathways that converge and interact to regulate mitochondrial energetics, biogenesis, ROS production, preservation, and repair of mitochondrial DNA (mtDNA), and mitophagy. Cellular senescence and mitochondrial dysfunction are closely linked: cell senescence, more specifically, persistent DDR signaling, directly contributes to senescence-associated mitochondrial dysfunction [25][99], while mitochondrial dysfunction drives and maintains cell senescence [27][70]. The combination of senescence and loss of mitochondrial homeostasis seems to have implications in maladaptive responses to cellular stress, increased vulnerability to injury, and the development of pulmonary fibrosis [42][57][28][71,94,100].
Impaired mitochondrial biogenesis in IPF potentially creates an imbalance in the production of cellular energy and the demand for it, resulting in mitochondrial dysfunction. NADPH oxidase 4 (Nox4), which is also known to be increased in senescent IFP myofibroblasts, can suppress mitochondrial biogenesis and bioenergetics via Nrf2 and the mitochondrial transcription factor Selective mitophagy of damaged mitochondria occurs through PINK1-Parkin signaling, with PINK1 acting as a sensor of mitochondrial membrane depolarization and subsequently activating Parkin, which labels the dysfunctional mitochondrion for trafficking to the autophagosome [29][105]. The fibroblast-to-myofibroblast differentiation mediated by TGF-β1 was characterized by reduced mitophagy and defects in mitochondrial function, at least in part due to PINK1 deficiency [31][106].
ROS can also damage lung epithelial cells and promote senescence of the surrounding cells [25][32][99,107]. mTOR activity also affects the signal transduction between mitochondria and the nucleus. The coordinated expression of mitochondrial and nuclear genes is necessary for maintaining the normal function of mitochondria and homeostatic levels of ROS. NOX4 can repress mitochondrial biogenesis in lung fibroblasts through the direct inhibition of Nrf2 and TFAM [33][104].
Indeed, diminished autophagy is observed during aging, and accelerated aging has been attributed to reduced autophagy [59][112]. In lung fibroblasts, Beclin1, LC3, and p62 have been described as autophagy-related biomarkers and demonstrate that there is decreased autophagy activity in lung tissues of IPF patients [60][61][114,115]. Beclin1, a key regulator of autophagy in IPF lung fibroblasts, is downregulated compared with normal lung fibroblasts [61][62][115,116]. Cellular senescence affects adaptive responses to stress, decreasing autophagy through the activation of mTORC1 in lung fibroblasts.
Autophagy is also involved in the regulation of the activated phenotype of IPF fibroblasts. An aberrant PTEN/Akt/mTOR axis desensitizes IPF fibroblasts from polymerized collagen-driven stress by suppressing autophagic activity, thereby producing a viable IPF fibroblast phenotype on collagen. This suggests that the aberrantly regulated autophagic pathway may play an important role in maintaining a pathological IPF fibroblast phenotype in response to a collagen-rich environment [63][118]. Deficient autophagy also seems to contribute to the invasive property of IPF fibroblasts [64][119].
In contrast to senescent epithelial cells, fibroblasts from IPF lungs are highly resistant to apoptosis [55][65][66][92,120,121]. The resistance to stress-induced cell death of senescent fibroblasts results in the persistence of “damaged” cells that would otherwise undergo apoptosis and be cleared from the site of wound repair [56][93]. Over time, this leads to the accumulation of senescent fibroblasts [16][56][41,93], which express high levels of αSMA and secrete excessive amounts of ECM components, promoting the development of fibrosis [58][95]. Further, little to no evidence of apoptosis was observed in α-SMA expressing cells in these areas, confirming the apoptotic resistant phenotype of senescent myofibroblasts in areas of lung fibrosis [56][93].
Perhaps the most studied have been the changes in the levels of Bcl-2 family proteins. Alterations in the expression of pro-apoptotic and antiapoptotic genes have also been associated with epigenetic modifications, including histone modification and DNA methylation. These site-specific histone modifications lead to active transcription of the Bcl-2 gene, which enhances apoptosis resistance in senescent fibroblasts [67][124]. The exogenous restoration of Sirt3 in aged mice promotes the activation of the forkhead box transcription factor FoxO3a in fibroblasts, the upregulation of pro-apoptotic proteins
Increased AKT activity is central to various signaling pathways involved in cell survival, and activation of the PI3K/AKT/mTOR pathway decreases autophagy and contributes to apoptosis resistance in IPF lung fibroblasts [68][117]. In contrast, a low activity of PTEN leading to the inactivation of the transcription activator FoxO3a through the PTEN/Akt-dependent pathway and downstream downregulation of caveolin-1 and Fas expression has also been shown [69][127]. Fas expression is necessary to sensitize lung fibroblasts to Fas ligation-induced apoptosis. The role of these pathways in apoptosis resistance in senescent fibroblasts and pulmonary fibrosis is evidenced by the mitigation of bleomycin-induced fibrosis in aged mice by quercetin via a reduction in AKT activation and upregulation of caveolin-1 and Fas levels [55][92].
The emergence of the senescent and apoptosis-resistant myofibroblast phenotype has also been attributed to an elevated expression of the ROS-generating enzyme Nox4 and an impaired capacity to induce the Nrf2 antioxidant responses [56][93]. Lung tissues from human subjects with IPF confirmed a high expression of Nox4 in fibroblastic foci, where Nrf2 expression is reduced. The in vivo knockdown of Nox4 and pharmacologic targeting of Nox4 during the persistent phase of lung fibrosis in aged mice reduced Bcl-2 levels and restored the capacity of senescent fibroblasts to undergo apoptosis, permitting fibrosis resolution.
These cells are enriched for pro-fibrotic and senescence factors and exhibit a global loss of transcripts encoding for components of various DNA damage response and repair proteins, including DNA-PKcs [70][130]. Several studies have previously shown the relationship between DNA damage and repair, senescence, and pulmonary fibrosis [71][132]. The loss of clusterin, for example, can promote disrepair and senescence in fibrotic lungs via the loss of DNA damage response and repair pathways [72][133]. Thus, a feedforward loop between the senescent environment, the expansion of MPCs and progeny, and the senescent and pathogenic behavior of these cells seem to be present in fibrotic lung diseases such as IPF.
However, the persistent or accumulation of senescent cells can certainly dysregulate the immune systems and transform the organ microenvironment to favor a chronic inflammatory state that, in part, is commonly observed in many age-related conditions, lung fibrosis included [73][74][75][76][138,139,140,141]. There are two characteristics of the immune system in the cellular senescence processes implicated in lung fibrosis: the dysfunction of the immune system called “immunosenescence”, and the senescence of immune cells per se [77][142]. Despite controversy in the role of inflammation in lung fibrosis due to the failure of many immunosuppressants in treating IPF [78][143], persistent chronic inflammation is undoubtedly one of the hallmarks of lung fibroproliferative disorders [78][143]. Here, we describe two main characteristics of immune dysfunction associated with cellular senescence in lung fibrosis.
Immunosenescence is broadly defined as declining immunity with age or prematurely with specific stimuli [79][80][81][144,145,146]. The main characteristics are a low proliferative index of the immune cells and maladaptive responses to triggers and stressors [82][83][84][85][147,148,149,150]. Although immunosenescence can occur irrespective of immune cell senescing [77][142], in most cases, it is the main consequence of immune cell senescence. Immunosenescence contributes to lung fibrosis pathogenesis in two ways: first, it promotes an accumulation of senescent cells that are the primary sources of SASP mediators [86][87][151,152], and second, it enhances proinflammatory mediators released from immune cells into the lung microenvironment, providing additive effects to SASP from other sources in reverberating inflammation.
Innate immunity is primarily driven by alveolar macrophages, natural killer (NK) cells, and dendritic cells (DC). The best characterization of innate immunosenescence is the alteration of the immune system in aging irrespective of the senescence of immune cells themselves [88][153]. An example of innate immune dysregulation is an increase in immature DC in the BAL and lungs of IPF patients [89][90][155,156]. Lastly, the decrease in matured CD57+cytotoxic NK cells and leukocytes in end-stage IPF is associated with defective immune surveillance necessary for senescent cell clearance, thereby promoting a profibrotic microenvironment [91][160].
A shift in the T cell and B cell population toward the memory phenotype and the progressive loss of the naïve cells are key features of adaptive immunosenescence [92][161]. For instance, an increase in Treg cells in aging promotes Th17 cell differentiation and IL-17 production in experimental lung fibrosis [93][94][163,164]. Similarly, IL-17A from T helper cells utilizes the IL-17A/IL-17RA axis to mediate rheumatoid arthritis-induced lung fibrosis [95][165]. The role of B cells in adaptive immunosenescence in lung fibrosis is much less studied.
The characterization of immune cell senescence is well-described in aging. However, this depiction is less straightforward in most lung fibrosis studies. Nonetheless, due to the intertwined nature of aging and lung fibrosis, it is conceivable that aged immune cells regulate lung fibrosis pathogenesis, in part, through the cellular senescence mechanism [85][150].
All of these changes cause a proinflammatory milieu but become ineffective in clearing senescent cells. The presence of p16Ink4a/β-galactosidase-positive macrophages in lung fibrosis resemble senescent macrophages seen in aging mice [96][171], which could explain senescent cell accumulation observed in lung fibrosis such that these profibrotic senescent macrophages lose their capacity to eliminate cytotoxic senescent cells. However, controversy arises surrounding the specificity of p16Ink4a/β-galactosidase positivity features, given that these macrophages did not undergo cell cycle arrest, as typically seen in senescent cells [97][172]. Lastly, aged macrophages are more hyperresponsive to noxious stimuli through NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3) inflammasome activation, demonstrating their profibrotic capacity [98][176].
The shift in T cell profiles from highly proliferative “naïve” T cells to steady “experienced” or effector memory T cells (CD25−CD45RA−CD45RO+CD127+) is the classic finding of T cell senescence in aging [99][177]. The standard molecular markers of senescent T cells are CD27, CD28, CD57, and Killer cell Functionally, the senescent T cells express higher levels of proinflammatory cytokines such as IFN-γ and TNF-α, or cytotoxic mediators such as granzyme B and perforin, which cause tissue damage and further activate other immune cells [100][179]. Although it is unclear whether targeting senescent T cells will improve clinical outcomes in lung fibrosis, they remain one of the promising therapeutic targets through alternative approaches.
Additional immune cells worth mentioning are circulating leukocytes, whereby the shortening of the telomere length of these cells was noted in familial and sporadic pulmonary fibrosis patients [101][184]. More recent evidence (in an abstract form) showed that SASP released by IPF senescent fibroblasts promoted the senescence of pulmonary NK cells that, in turn, permitted the accumulation of senescent fibroblasts, thereby creating a vicious cycle of persistent chronic inflammation [102][97]. Specifically, CD3+CD4+CD25highFoxp-3+cells were elevated in IPF, whereas Th17 cells were significantly compromised [103][185].
In summary, despite the debate surrounding the contribution of chronic inflammation in lung fibrosis pathogenesis, more direct and indirect evidence supports the roles of the senescent state of the immune response. The intertwined immune dysregulation in cellular senescence is the basic principle of senescence pathogenic mechanisms. In addition, impaired immune surveillance is responsible for the accumulation of senescent cells in aging lungs [104][188]. The continual accretion of these senescence cells, in turn, elevates SASP mediator levels in the fibrotic foci that can further recruit immune cells, promoting a chronic persistent inflammatory state and the progression of fibrosis [74][105][139,189].
MSCs, also known as mesenchymal stromal cells, are a population of adult stem cells that were first described in the bone marrow but have been described in many tissues. These cells are multipotent stromal cells that can differentiate into a variety of cell types and, thus, have an important role in tissue remodeling and repair [106][190]. In silica-induced lung fibrosis in mice, intravenous administration of B-MSCs ameliorated lung fibrosis in that B-MSCs homed to injured lungs and replenished the injured epithelial cells [107][191]. B-MSCs of IPF patients displayed senescence phenotypic changes, including the decreased replication rate, upregulation of common senescence markers, and the presence of DNA damage.
In summary, known key cellular players in lung fibrosis pathogenesis, i.e., epithelial cells, mesenchymal cells, and immune cells exhibit cellular senescence phenotypes in pre-clinical studies and in human lung specimens. It further emphasizes the critical role of the senescence regulatory pathway. Although different studies highlight their cells of interest, it is likely that senescence processes are present in more than one cell type in the lung, as has been demonstrated in robust single-cell RNA sequencing studies. It is unclear at this stage whether these senescent cells work in concert to promote lung fibrosis, or whether one dominant cell type drives the process.