The Homologous Recombination Deficiency (HRD) phenotype makes these tumors sensitive to DNA double strand break-inducing agents, including poly-(ADP-ribose)-polymerase (PARP) inhibitors.
- HRD
- BRCAness
- DNA repair
1. Homologous Recombination Deficiency and Cancer
The discovery of new biomarkers of tumor treatment response is the focus of many studies. Several markers have made it to the clinic, such as hormone receptor status for breast and prostate cancer. However, most anti-cancer drugs are prescribed based on overall efficacy in particular tumor types without preselection based on predictive biomarkers. Developing biomarkers to select the best treatment for each patient should increase patient prognosis and decrease unnecessary side effects and costs.
Here, we describe methods to select patients who might benefit specifically from a specific class of DNA damaging agents that induce double strand DNA breaks (DSB), including platinum salts and poly-(ADP-Ribose)-polymerase inhibitors (PARPi). The DSB caused by these agents require homologous recombination (HR) for their repair [1,2][1][2]. Patients with a germline mutation in the breast cancer susceptibility genes Breast cancer 1 (BRCA1) or Breast cancer 2 (BRCA2) have a high risk of developing breast cancer or ovarian cancer [3] [3] and show high sensitivity to these DNA damaging agents [4,5][4][5]. As both BRCA1 and BRCA2 proteins are involved in HR [66][7],7], tumors without BRCA gene mutations but with a similar homologous recombination deficiency (HRD) phenotype are expected to show similar responses to DSB-inducing drugs. Currently, germline BRCA (gBRCA) mutated ovarian and breast cancer patients are eligible for PARPi treatment, but a clinically validated test to select other HRD tumors (also called BRCAness or BRCA-like tumors) is not yet available.
HR mediates error-free repair of DSBs that arise during DNA replication [8]. BRCA1 is attracted to DNA ends, where it stimulates exonucleases that create a single strand (ss) overhang by 5′-3′ end resection (Figure 1). RPA binds to the ssDNA and is subsequently replaced by RAD51 protein. This exchange reaction requires a complex containing RAD51, BRCA2, and PALB2. After formation of the RAD51-DNA filament, the single strand overhang searches for homology in the sister chromatid, that functions as the template for DNA repair. As HR uses the newly formed sister chromatid as a template, it is restricted to the S and G2 phases of the cell cycle [7].
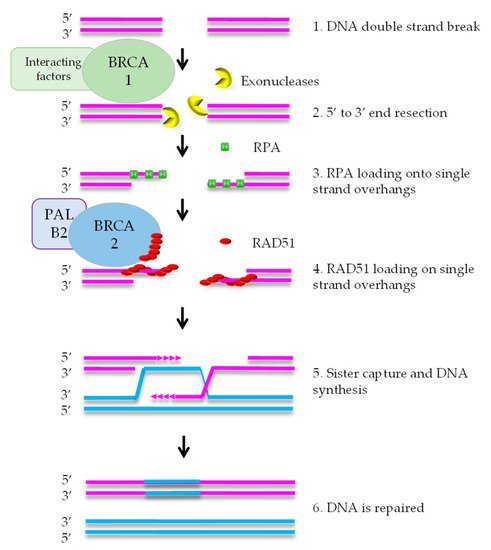
DNA repair defects have been described as ‘enabling characteristics of cancer’ [9]. This means that defects such as HRD promote tumorigenesis by increasing the speed of mutation accumulation in a cell on its way to become a cancer cell. The DNA repair defect is often an early event in tumor evolution and the defect is present in the large majority, if not all, of the cells in the tumor. Therefore, HRD is a promising therapeutic target.
2. Various ways to Detect HRD in Tumors
In the search for BRCAness, several tests have been designed to recognize patients with a HRD phenotype. Many tests are based on sequencing. The most obvious test is sequencing of known disease causing genes, i.e. BRCA1 or BRCA2. As an extension of this approach, panels of multiple genes can be chosen for example based on their involvement in the HR pathway. The PALB2 gene is an example of a HR gene that predicts PARPi sensitivity, but many other HR genes are also expected to have predictive value. It is currently not clear which additional genes should be included in such sequencing panels.
Alternative tests are based on loss and amplification of pieces of chromosomes or whole chromosomes. The basic idea behind these tests is that HRD is often accompanied by very specific chromosomal alterations, which give specific growth advantages to HRD tumors. For example, loss of function of the TP53 gene prevents apoptosis caused by inactivation of HR which results in replication stress. Such a pattern of chromosomal changes can therefore be used as a way to identify HRD tumors.
Finally, sequencing can also be used in the search for specific types of genomic changes that are a direct or indirect consequence of the DNA repair defect, so-called scars, which accumulate over the course of tumor evolution. BRCA-deficient tumors accumulate specific patterns of point mutations, as well as specific types of DSB repair events. A very characteristic type of repair event is rejoining of DSB with microhomologies at the junctions. Microhomologies are short stretches (generally 1-6 base pairs) of directly repeated nucleotides. Therefore, this type of repair causes loss of the intervening sequence. Detecting these sequences requires Whole Genome Sequencing (WGS), which is currently rather expensive. However, it is expected that new technology will make this approach affordable on a routine basis in the future.
A completely different approach directly investigates functional parameters in fresh tumor specimens. These so-called functional assays use material from the tumor to test a relevant treatment, followed by measuring the response of these living tumor specimens to assess whether the tumor harbors deficiencies in a relevant process. HRD can be identified by testing accumulation of the RAD51 protein on DSB in a small piece of tumor material. A major advantage of this approach is that it measures directly whether the tumor has a defect without assumptions about the genes involved or correlated consequences of the repair defect. However, the use of fresh tumor material requires specialized laboratory adaptations, which may be challenging in hospital settings.
These various ways of selecting patients have been described in more detail in a recent review, which also summarizes evidence for their analytical validity, clinical validity and clinical utility.
References
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817.
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294.
- Chen, S.; Parmigiani, G. Meta-analysis of brca1 and brca2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333.
- Mylavarapu, S.; Das, A.; Roy, M. Role of brca mutations in the modulation of response to platinum therapy. Front. Oncol. 2018, 8, 16.
- Keung, M.Y.T.; Wu, Y.; Vadgama, J.V. Parp inhibitors as a therapeutic agent for homologous recombination deficiency in breast cancers. J. Clin. Med. 2019, 8, 435.
- Carvalho, J.F.; Kanaar, R. Targeting homologous recombination-mediated DNA repair in cancer. Expert Opin. Ther. Targets 2014, 18, 427–458.
- Wyman, C.; Kanaar, R. DNA double-strand break repair: All’s well that ends well. Annu. Rev. Genet. 2006, 40, 363–383.
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.