1. Introduction
The ubiquitin-proteasome system (UPS) represents a crucial cellular mechanism for highly regulated proteolysis and protein quality control process in eukaryotes
[1][2][1,2]. The 26S proteasome is a large multi-subunit protease of ~2.5 MDa for selective degradation of intracellular proteins that are tagged by ubiquitins
[3][4][3,4]. Recent findings indicate that proteasome is actively adapted to a large network of protein interactions for discrete degradation events, and such adaptability may also be controlled through a multitude of proteasome’s conformational transitions
[5][6][7][8][9][5,6,7,8,9]. Notably, deubiquitinases (DUBs), which exclusively reverse the ubiquitination process in the UPS, are also critically associated with the proteasome
[10][11][12][10,11,12]. In mammals, the regulatory particle (RP) of the 26S proteasome contains three major classes of DUBs–USP14 (Ubp6 in budding yeast), RPN11 (also known as PSMD14), and UCH37 (also known as UCH-L5) ()
[3][11][12][13][3,11,12,13]. USP14/Ubp6 is a reversible interactor with the proteasome, and its activity can be highly enhanced by association with the proteasome
[12][14][15][16][12,14,15,16]. USP14 is capable of sparing the substrates from degradation prior to the proteasome’s commitment step and shows remarkable preference for multi-chain bearing ubiquitin conjugates
[16][17][18][16,17,18]. By contrast, RPN11 is an integral subunit of the proteasome, and this metalloprotease is essentially coupled to substrate degradation in an ATP-dependent manner
[11][19][20][11,19,20]. Although USP14 and RPN11 may mediate opposite proteolytic consequences, both of the enzymes apparently share a similar en bloc or proximal cleavage mechanism
[11][17][11,17]. The function of UCH37 on the proteasome remains to be further established because this DUB may distally trim the ubiquitin chains for rescuing the substrates from degradation but also can selectively debranch the K48-linkage among a complex mixture of bifurcate ubiquitin conjugates for enhanced substrate degradation
[12][21][22][12,21,22]. DUBs are emerging as attractive therapeutic targets because they may control the turnover rate of a number of intracellular proteins, including ones that might be highly deregulated in the disease states
[23][24][23,24]. The isopeptidase activities of DUBs can be selectively inhibited by catalytic site-directed drug-like compounds. Moreover, recent advances in developing robust screening technologies with more refined chemical libraries have successfully yielded promising small-molecule DUB antagonists of active site-directed inhibitors as well as allosteric inhibitors
[24][25][26][27][24,25,26,27]. Specific DUB inhibition on the proteasome is particularly appealing because each proteasome-associated DUB can exert distinct influence over the proteolytic outputs (A). Therefore, it is not surprising that considerable efforts from academia and industry have also been put towards developing drug-like molecules for targeting proteasome-associated DUB activities
[23][24][28][23,24,28]. Such specific DUB inhibitors at the proteasome not only offer exciting degradation-based therapeutic strategies but also serve as valuable chemical tools to reveal novel deubiquitination biology for dynamic proteasome function.
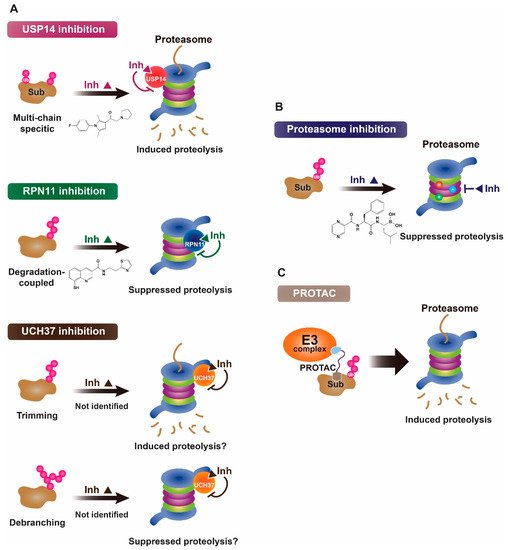
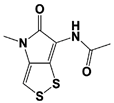
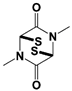
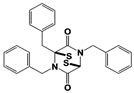
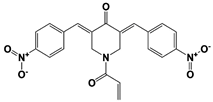
Figure 1. Proposed working mechanisms of proteasomal deubiquitinase inhibitors and their comparison to proteasome inhibitor and PROTAC. (A) (Top) USP14’s multi-chain specific cleavage activity can be selectively targeted by USP14 inhibitors (e.g., IU1 is shown as an example), resulting in induced degradation of substrates. (Middle) Degradation-coupled RPN11 activity can be selectively inhibited such as by capzimin as shown. RPN11 inhibition can strongly suppress the proteasome-mediated substrate degradation. (Bottom) UCH37 specific inhibitors–which have not been developed yet–may exert differential effects on proteolysis depending on the type of ubiquitin conjugates. Unbranched or poorly ubiquitinated substrates might be highly subject to UCH37’s trimming activity, and its specific inhibition may lead to induced protein degradation. By contrast, degradation of branched ubiquitin conjugates is likely to be attenuated by UCH37 inhibition. (B,C) Proteasome inhibitor (e.g., bortezomib as shown) and PROTAC are depicted as examples of proteolysis suppressor and inducer, respectively. Color-coded circles in proteasome at B indicate each pair of proteasome’s active sites. PROTAC is a chimeric compound closely linking E3 and target substrate, thus facilitating the ubiquitination process. Inh, inhibitor. See the text for more details.
2. Proteasomal Deubiquitinases as Therapeutic Targets
For the past decades, the UPS has been clearly recognized among the most important drug targets because of its critical contribution to protein homeostasis, signaling pathways, and cellular physiology; its deregulation or genetic alteration is intimately associated with human pathogenesis
[29][30][31][32][33][29,30,31,32,33]. The success story of proteasome inhibitors for cancer therapy highlights the clinical importance of the UPS as valid targets that can be even further expanded into various aspects of the proteolytic system and other types of pathophysiology
[34][35][34,35]. In fact, a recently emerging novel paradigm of “induced proteolysis”, such as by PROteolysis TArgeting Chimera (PROTAC), can chemically harness the endogenous ubiquitination machinery for targeted protein degradation (B)
[36][37][36,37].
As opposed to proteasome inhibition, this new concept defines the UPS as yet another class of extraordinary drug target for effectively disposing of the conventionally intractable or “undruggable” disease-associated proteins ()
[24][38][24,38]. In the similar context, deubiquitylation reactions may offer exciting opportunities for developing promising drug candidates due to their key roles in the proteolytic pathways as well as other biological processes
[23][24][23,24]. Although the development of specific DUB inhibitors is challenging per se and still in its early stage, recently performed a series of elegant works have produced nice examples of highly selective small-molecule inhibitors for targeting USP7 and USP30
[24][39][40][41][42][43][44][24,39,40,41,42,43,44].
Targeting DUBs on the proteasome may also represent unique therapeutic strategies for actively regulating the proteasome-mediated proteolysis in a dynamic manner. Individual or ensemble of deubiquitination activities can exert distinct and multiple impacts on the proteasome before or throughout substrate processing (A); such DUB-imposed regulation may render the proteasomal activities to be highly versatile, and in this sense, the proteasome acts as a critical hub as well as a rate limiting step for the ubiquitin-dependent degradation pathways
[11][12][11,12]. Recent high resolution cryo-electron microscopy (cryo-EM) studies also have identified a number of conformational states of substrate-free and substrate-bound proteasomes, in which the proteasome-associated DUBs are likely to actively and differentially modulate the degradation events by sensing those conformational dynamics
[4][5][12][4,5,12].
Among three major proteasomal DUBs, USP14 or its yeast ortholog Ubp6 is a thiol protease that is only transiently associated with the proteasome; thus, this enzyme may favor the specific conformational states of the proteasome
[14][15][16][45][46][47][14,15,16,45,46,47]. Earlier genetic studies have revealed that USP14/Ubp6 is a sensitive responder to ubiquitin and proteasome stress, and also to proteotoxic stress, although in general its deficiency is tolerable for cell survival
[48][49][50][51][52][53][54][48,49,50,51,52,53,54]. During the mouse development, however, this isopeptidase is critically involved in the motor neuron function partially through the noncatalytic mechanism
[55][56][57][58][55,56,57,58]. As a therapeutic target, USP14 has been best studied in neurological disorders and cancers
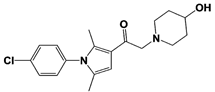
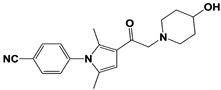
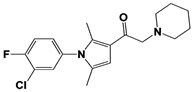
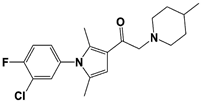
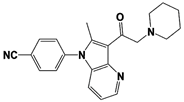
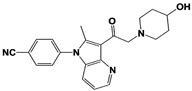
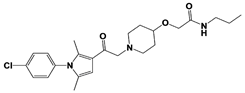
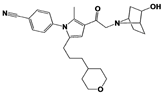
[23][59][23,59]. USP14 and its inhibitors (as discussed in
Section 3) have been reported to regulate several pathological targets, such as Tau, ATXN3, TDP-43, GFAP, and PrP that are highly implicated in neurodegenerative diseases
[16][60][61][62][16,60,61,62]. Intriguingly, apart from its inhibitory role on the proteasome, USP14 was also found to negatively regulate autophagy in mammalian cells and basal mitophagy in fly models
[63][64][63,64]. USP14 expression is upregulated in several cancers including lung adenocarcinoma, ovarian cancers, esophageal squamous cell carcinoma, and pancreatic ductal adenocarcinoma, in which this enzyme is often positively correlated with tumor recurrence, metastasis, and poor patient survival
[65][66][67][68][69][65,66,67,68,69]. Albeit seemingly a promising clinical target, the underlying mechanisms of how USP14 participates in those disease processes still remain to be elucidated.
In contrast to USP14, RPN11/PSMD14 (also known as POH1) is an essential subunit of the proteasome that belongs to JAMM/MPN metalloprotease class of DUB
[19][20][70][19,20,70]. Recent cryo-EM studies provide the structural basis of how RPN11′s DUB activity can be coupled to substrate translocation and degradation in an ATP-dependent fashion
[6][11][13][47][71][72][73][6,11,13,47,71,72,73]. Along with other RP components, this metalloprotease undergoes noticeable structural changes during the transitions from the substrate-free state to the substrate-processing states of the proteasome. This conformational switch drives RPN11 to be catalytically productive for the committed substrates in both repositioning on the proteasome and reshaping the local structure of its featured Ins-1 loop. Due to its strict requirement for proteasomal degradation, the genetic depletion or catalytic mutation of RPN11 causes the lethality
[19][49][51][74][75][76][19,49,51,74,75,76]. Therefore, the successful targeting strategy for cancer therapy by proteasome inhibitors might be also similarly applied to RPN11-mediated inhibition of proteasomal degradation
[77]. The key difference here, however, is that in contrast to the core particle (CP)-directed catalytic inhibition, RPN11 inhibition will occur on the RP, and thus is likely to show more specific effects. Besides, RPN11 has been implicated in oncogenesis as a potential drug target; its expression level is positively correlated with tumor formation and metastasis, while the genetic depletion or pharmacological inhibition showing the opposite effects–such as in hepatocellular carcinoma, multiple myeloma, breast cancer, esophageal cancer, colorectal cancer, and prostate cancer
[78][79][80][81][82][83][78,79,80,81,82,83].
Like USP14, UCH37/UCH-L5 is a thiol protease class of DUB that is reversibly associated with the 19S RP of the proteasome; its binding is mediated by RPN13/ADRM1, a ubiquitin receptor which can also markedly enhance the UCH37′s activity
[84][85][86][84,85,86]. An intriguing feature of UCH37 is that this enzyme belongs to both the proteasome and the INO80 chromatin-remodeling complex in a mutually exclusive manner; its DUB activity can be selectively activated only when bound to the proteasome
[87][88][89][87,88,89]. UCH37 was reported to trim the distal ubiquitin from erroneously ubiquitinated proteins for their rescue
[21], or it does so to release proteasome-occupying unanchored chains for the productive round of substrate loading
[90]. Interestingly, a recent study demonstrated that UCH37 on the proteasome can selectively cleave the K48-linked branched chains to promote the degradation of substrates
[12][22][12,22]. In any case, the exact physiological functions of UCH37 remain largely elusive. Like USP14 and RPN11, several lines of studies have reported that UCH37 expression is elevated in a number of cancers including esophageal squamous cell carcinoma, hepatocellular carcinoma, epithelial ovarian cancer, endometrial cancer, and lung adenocarcinoma, in which this protease is associated with tumor progression and poor patient survival
[91][92][93][94][95][96][91,92,93,94,95,96].