In our review, we provide an overview of the molecular effects of traditional and more recently FDA-approved Multiple Sclerosis (MS) drugs on four CNS cell types.
- fingolimod
- dimethyl fumarate
- teriflunomide
- glatiramer acetate
- interferon-β
- microglia
- astrocyte
- neuron
- oligodendrocyte
- multiple sclerosis drug action
Introduction
MS is an inflammatory disease of the central nervous system (CNS) characterized by oligodendrocyte pathology, microgliosis, astrogliosis, alterations of the blood–brain barrier (BBB), demyelination and neurodegeneration, and an exacerbating infiltration of both innate and adaptive immune cells into the brain [1][2]. MS is a complex disease with a large heterogeneity in MS lesions [3][4]. Furthermore, the non-lesioned white- and grey-matter regions in MS brains are different from those in healthy individuals [2][3]. For quite some time, the dysregulation of the peripheral immune system, causing immune cells infiltrating the CNS, autoreactivity against myelin sheath components and secondary BBB dysfunction, has been considered to be the primary cause of MS CNS pathology, defined as the outside-in hypothesis [5]. However, more recent research on MS and other neurodegenerative diseases has indicated a central role for a distinct type of macrophage found in the CNS, the microglia [6][7]. The hypothesis in which MS pathology is first and foremost caused by CNS-intrinsic factors, subsequently leading to the infiltration of peripheral immune cells via a leaking BBB, represents the inside-out model [8][9], which is supported by pathological evidence showing the absence of peripheral immune cells in newly forming MS lesions [10].
Because the outside-in model has been the norm for a long time, the currently available MS drugs approved by the Food and Drug Administration (FDA) have been mainly designed to target various cell types within the peripheral immune system [11] and most drug-impact studies have been directed towards their peripheral effects on the cells of the adaptive immune system [12]. However, it is likely that the MS drugs also affect (innate) CNS cells and the molecular cascades associated with neuroinflammation, since most genes that are dysregulated in MS-peripheral immune cells are also expressed in microglia [13]. MS drug effects on CNS pathology have been mostly studied in humans and animals on the basis of the clinical features of disease progression, magnetic resonance imaging (MRI) measures, and blood or cerebrospinal fluid (CSF) levels of biomarkers for demyelination and neuronal degeneration [14][15][16]. For this reason, we set out to review studies assessing at the molecular level, the effects of MS drugs on the pathways operational in CNS cells.
Molecular effects on cell types in the CNS have been reviewed for a number of FDA-approved MS drugs, such as Fingolimod (FTY720; Gilenya), Dimethyl Fumarate (DMF; Tecfidera), Glatiramer Acetate (GA; Copaxone), Interferon-beta (IFN-β; Rebif, Avonex, Betaseron, Extavia, Plegridy) and Teriflunomide (TF; Aubagio) [17][18][19][20][21][22][23][24][25][26][27][28]. The CNS-directed molecular effects of more recently approved drugs, such as Laquinimod (LQ; Nerventra), Natalizumab (NZ; Tysabri), Alemtuzumab (AZ; Lemtrada) and Orcelizumab (OCR; Ocrevus), have been less well described, except for the neuroprotective effects of LQ and NZ [29][30][31]. In general, each of these previous studies has reported the (molecular) effects of only one or two MS drugs (e.g., [28][29][31]) on one or two CNS cell types (e.g., [22]). Moreover, the protective effects of MS drugs on neurons and oligodendrocytes have often been attributed to indirect effects caused by the actions of MS drugs on peripheral immune cells (e.g., [28]). Therefore, the effects of MS drugs have not been documented in multiple CNS cell types nor integrated into a common molecular cascade of events. The goal of the present review is to describe and compare the molecular effects of the traditional and recent FDA-approved MS drugs on multiple CNS cell types, focusing on microglia within the generally applied homeostatic (M0), pro-inflammatory (M1) and anti-inflammatory (M2) designation [32][33], and on astrocytes within the homeostatic (A0), reactive (A1) and neuroprotective (A2) nomenclature [34], as well as on neurons and oligodendrocytes.
Main results
Except for IFN-β, the various FDA-approved MS drugs described in the review have a clear effect on the transition from a pro-inflammatory M1 into an anti-inflammatory M2 microglia phenotype. This effect is particularly evident in inflammatory (EAE) as well as non-inflammatory (Cuprizone, Lysolecithin, TBI, AD) rodent models in which neurotoxicity and demyelination are central factors causing pathology. The results of the in vitro studies are in line with this inference. Moreover, most FDA-approved MS drugs reduce the inflammatory environment in the CNS by converting reactive A1 astrocytes into a neuroprotective A2 phenotype. Comparable to what holds for microglia, this effect was evident from in vitro cellular studies as well as from studies on wild-type rodents, rodent CNS-related disease models and MS patients.
Central to the mechanism of action of the FDA-approved MS drugs appeared to be NFκB signaling. NFκB is the major signal transducer involved in the activation of microglia and astrocytes towards a pro-inflammatory M1 and reactive A1 phenotype, respectively [35]. This transcription factor complex, that binds to nuclear DNA elements, is responsible for activating the transcription of a wide range of pro-inflammatory cytokines, chemokines and matrix metalloproteinases, and induces oxidative stress and inflammasome activation [36] [37]. Moreover, NFκB interacts and cooperates with two other nuclear signal transducers, STAT1 and STAT3 [38][39][40]. A number of studies has described an increased degree of phosphorylation of NFκB, STAT1 and STAT3 under various CNS disease and injury conditions, linking their activation to the induction of a number of pathological states [41][42][43][44]. The effects of the MS drugs on pro-inflammatory as well as anti-inflammatory factors, such as chemokines, growth factors and oxidative stress inducers that are transcriptionally driven by NFκB, strongly indicate that in microglia the NFκB pathway plays a key role in the molecular actions of these drugs. Moreover, in microglia both FTY720 and DMF reduce the protein levels and activation of NFκB [45][46][47][48][49][50][51][52], and virtually all MS drugs modulate the expression of a remarkable number of molecular effectors upstream of NFκB, such as MAPK and PI3K/AKT [34][47][53][52][54], and other proteins known to influence NFκB signaling. Similarly, in astrocytes most MS drugs discussed here affect the expression of pro-inflammatory and anti-inflammatory factors that is dependent on NFκB-induced transcriptional programs. In addition, in astrocytes FTY720 and LQ diminish NFκB protein levels and activation [55][56][57][58][59][60][61][62][63], and nearly all of the MS drugs modulate the expression of MAPK and ERK1/2 [64][65][66] as well as of other NFκB-interactor proteins. Together, these findings highlight the importance of the NFκB pathway for MS drug actions in microglia as well as astrocytes. A summary of these results is depicted in Figure 1. In this connection, one should realize that the effects of the monoclonal antibodies NZ, AZ and OCR on CNS cells have not yet been sufficiently studied. Furthermore, IFN-β seems to stimulate rather than inhibit the pro-inflammatory NFκB and STAT1 pathways [67][68][69][70][71], and the STAT1-related kinases JAK1 and TYK1 [70] in microglia and astrocytes [68][71][72][73].
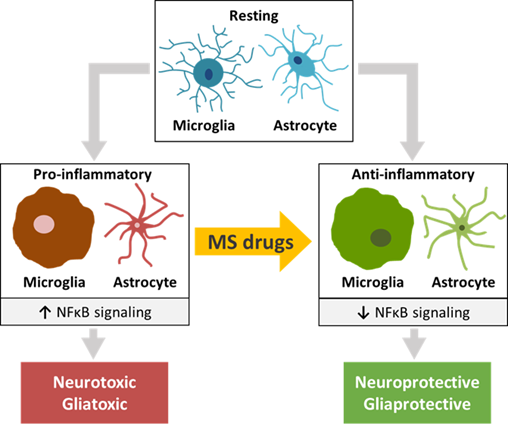
Figure 1. The effects of traditional and more recent FDA-approved MS drugs may be partially attributed to their influence on glial cells and neurons of the CNS. A common molecular effect of these drugs may involve NFκB signaling, causing a switch from pro-inflammatory microglia and astrocytes to anti-inflammatory phenotypes of these CNS cell types that recently emerged as central players in MS pathogenesis. The switch may have a beneficial effect on the functioning of diseased or injured neurons and oligodendrocytes.
Conclusions
We provide an overview of the molecular effects of traditional and more recently FDA-approved MS drugs on four CNS cell types. The effects of these MS drugs on the peripheral immune system and their influence on immune cell infiltration via the BBB have been documented before, and were not specifically addressed in the review. From our comprehensive analysis of MS drug effects on CNS cells, we concluded that, via NFκB signaling, the majority of these drugs attenuates the pro-inflammatory M1 microglia and reactive A1 astrocytic phenotypes. On its turn, this attenuation may positively affect the functioning of diseased or injured neurons and oligodendrocytes. Such a mechanism is likely more complicated than initially thought in that microglia have recently been shown to engage a large range of activation programs which appear to be increasingly difficult to classify as purely pro-inflammatory versus anti-inflammatory [34][74]. Future characterization of the signatures of the recently described additional microglia and astrocytic subtypes, and of the extent of their pro-inflammatory or anti-inflammatory state, will aid in obtaining an even more detailed molecular understanding of the CNS mechanisms of action of drugs targeting MS. Knowledge of these molecular mechanisms may help anticipating adverse drug effects and in considering the use of combinatorial drug therapy to treat this complex neuroinflammatory disease.
References
- Hans Lassman; Multiple Sclerosis Pathology. Cold Spring Harb Perspect Med 2018, 8(3), a028936, DOI: 10.1101/cshperspect.a028936.
- Alexandra Kutzelnigg; Claudia F. Lucchinetti; Christine Stadelmann; W. Brück; Helmut Rauschka; Markus Bergmann; Manfred Schmidbauer; Joseph E. Parisi; Hans Lassmann; Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain 2005, 128, 2705-2712, 10.1093/brain/awh641.
- Tobias Zrzavy; Simon Hametner; Isabella Wimmer; Oleg Butovsky; Howard L. Weiner; Hans Lassmann; Loss of 'homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900-1913, 10.1093/brain/awx113.
- Hans Lassmann; Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Frontiers in Immunology 2019, 9, 3116, 10.3389/fimmu.2018.03116.
- Clare Baecher-Allan; Belinda J Kaskow; Howard L Weiner; Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768, DOI: 10.1016/j.neuron.2018.01.021.
- Sara Bachiller; Itzia Jimenez Ferrer; Agnes Paulus; Yiyi Yang; Maria Swanberg; Tomas Deierborg; Antonio Boza-Serrano; Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Frontiers in Cellular Neuroscience 2018, 12, 488, 10.3389/fncel.2018.00488.
- Dafina M. Angelova; David Ronald Brown; Microglia and the aging brain: are senescent microglia the key to neurodegeneration?. Journal of Neurochemistry 2019, 151, 676-688, 10.1111/jnc.14860.
- Peter K. Stys; Gerald W. Zamponi; Jan Van Minnen; Jeroen J. G. Geurts; Will the real multiple sclerosis please stand up?. Nature Reviews Neuroscience 2012, 13, 507-514, 10.1038/nrn3275.
- Maria Traka; Joseph R Podojil; Derrick P McCarthy; Stephen D. Miller; Brian Popko; Oligodendrocyte death results in immune-mediated CNS demyelination. Nature Neuroscience 2015, 19, 65-74, 10.1038/nn.4193.
- M H Barnett; John W. Prineas; Relapsing and remitting multiple sclerosis: Pathology of the newly forming lesion. Annals of Neurology 2004, 55, 458-468, 10.1002/ana.20016.
- Mehrdad Gholamzad; Masoumeh Ebtekar; Mehdi Shafiee Ardestani; Maryam Azimi; Zeinab Mahmodi; Mohammad Javad Mousavi; Saeed Aslani; A comprehensive review on the treatment approaches of multiple sclerosis: currently and in the future. Inflammation Research 2018, 68, 25-38, 10.1007/s00011-018-1185-0.
- Verena Loleit; Viola Biberacher; B Hemmer; Current and future therapies targeting the immune system in multiple sclerosis.. Current Pharmaceutical Biotechnology 2014, 15, 276-296, 10.2174/1389201015666140617104332.
- International Multiple Sclerosis Genetics Consortium*†; Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188, 10.1126/science.aav7188.
- Christiane Graetz; Sergiu Groppa; Frauke Zipp; Nelly Siller; Preservation of neuronal function as measured by clinical and MRI endpoints in relapsing-remitting multiple sclerosis: how effective are current treatment strategies?. Expert Review of Neurotherapeutics 2018, 18, 203-219, 10.1080/14737175.2018.1438190.
- Jens Kuhle; Harald Kropshofer; Dieter A. Haering; Uma Kundu; Rolf Meinert; Christian Barro; Frank Dahlke; Davorka Tomic; David Leppert; Ludwig Kappos; et al. Blood neurofilament light chain as a biomarker of MS disease activity and treatment response. Neurology 2019, 92, e1007-e1015, 10.1212/wnl.0000000000007032.
- on behalf of the MAGNIMS study group; Mike P. Wattjes; Álex Rovira; David Miller; Tarek A. Yousry; Maria P. Sormani; Nicola De Stefano; Mar Tintoré; Cristina Auger; Carmen Tur; et al.Massimo FilippiMaria A RoccaFranz FazekasLudwig KapposChris PolmanFrederik BarkhofX. Montalban MAGNIMS consensus guidelines on the use of MRI in multiple sclerosis—establishing disease prognosis and monitoring patients. Nature Reviews Neurology 2015, 11, 597-606, 10.1038/nrneurol.2015.157.
- Yasuyuki Kihara; Systematic Understanding of Bioactive Lipids in Neuro-Immune Interactions: Lessons from an Animal Model of Multiple Sclerosis.. Advances in Experimental Medicine and Biology 2019, 1161, 133-148, 10.1007/978-3-030-21735-8_13.
- Kristina Candido; Henry Soufi; Mausumi Bandyopadhyay; Subhajit Dasgupta; Henry Sioufi; Therapeutic Impact of Sphingosine 1-phosphate Receptor Signaling in Multiple Sclerosis. Mini-Reviews in Medicinal Chemistry 2016, 15, 1-8, 10.2174/1389557515666150709122517.
- Aran Groves; Yasuyuki Kihara; Jerold Chun; Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy.. Journal of the Neurological Sciences 2013, 328, 9-18, 10.1016/j.jns.2013.02.011.
- Lawrence G. Miller; Jennifer A. Young; Swapan K. Ray; Guanghu Wang; Sharad Purohit; Naren L. Banik; Somsankar Dasgupta; Sphingosine Toxicity in EAE and MS: Evidence for Ceramide Generation via Serine-Palmitoyltransferase Activation. Neurochemical Research 2017, 42, 2755-2768, 10.1007/s11064-017-2280-2.
- Jürgen Brück; Ralf Dringen; Adriana Amasuno; Ignasi Pau-Charles; Kamran Ghoreschi; A review of the mechanisms of action of dimethylfumarate in the treatment of psoriasis. Experimental Dermatology 2018, 27, 611-624, 10.1111/exd.13548.
- Azadeh Yazdi; Maryam Ghasemi-Kasman; Mohammad Javan; Possible regenerative effects of fingolimod (FTY720) in multiple sclerosis disease: An overview on remyelination process. Journal of Neuroscience Research 2019, 98, 524-536, 10.1002/jnr.24509.
- Samuel F. Hunter; James D. Bowen; Anthony T. Reder; The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis.. CNS Drugs 2016, 30, 135-47, 10.1007/s40263-015-0297-0.
- Jeffrey Liddell; Are Astrocytes the Predominant Cell Type for Activation of Nrf2 in Aging and Neurodegeneration?. Antioxidants 2017, 6, 65, 10.3390/antiox6030065.
- Jing Chen-Roetling; Raymond F. Regan; Targeting the Nrf2-Heme Oxygenase-1 Axis after Intracerebral Hemorrhage. Current Pharmaceutical Design 2017, 23, 2226-2237, 10.2174/1381612822666161027150616.
- Roland Liblau; Glatiramer acetate for the treatment of multiple sclerosis: evidence for a dual anti-inflammatory and neuroprotective role. Journal of the Neurological Sciences 2009, 287, S17-S23, 10.1016/s0022-510x(09)71296-1.
- Ruth Arnon; Rina Aharoni; Neurogenesis and Neuroprotection in the CNS — Fundamental Elements in the Effect of Glatiramer Acetate on Treatment of Autoimmune Neurological Disorders. Molecular Neurobiology 2007, 36, 245-253, 10.1007/s12035-007-8002-z.
- Suhayl Dhib-Jalbut; Mechanisms of action of interferons and glatiramer acetate in multiple sclerosis.. Neurology 2002, 58, S3-S9, 10.1212/wnl.58.8_suppl_4.s3.
- Finn Sellebjerg; Diego Cadavid; Deborah Steiner; Luisa M Villar; Richard Reynolds; Daniel Mikol; Exploring potential mechanisms of action of natalizumab in secondary progressive multiple sclerosis. Therapeutic Advances in Neurological Disorders 2015, 9, 31-43, 10.1177/1756285615615257.
- B. C. Kieseier; Defining a role for laquinimod in multiple sclerosis. Therapeutic Advances in Neurological Disorders 2014, 7, 195-205, 10.1177/1756285614529615.
- W. Brück; C. Wegner; Insight into the mechanism of laquinimod action. Journal of the Neurological Sciences 2011, 306, 173-179, 10.1016/j.jns.2011.02.019.
- Ruben Orihuela; Christopher A McPherson; G. Jean Harry; Microglial M1/M2 polarization and metabolic states. British Journal of Pharmacology 2015, 173, 649-665, 10.1111/bph.13139.
- Yu Tang; Weidong Le; Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Molecular Neurobiology 2015, 53, 1181-1194, 10.1007/s12035-014-9070-5.
- Sean J. Miller; Astrocyte Heterogeneity in the Adult Central Nervous System. Frontiers in Cellular Neuroscience 2018, 12, 401, 10.3389/fncel.2018.00401.
- Piotr Przanowski; Michal Dabrowski; Aleksandra Ellert-Miklaszewska; Michal Kloss; Jakub Mieczkowski; Beata Kaza; Anna Ronowicz; Feng Hu; Arkadiusz Piotrowski; Helmut Kettenmann; et al.Jan KomorowskiBozena Kaminska The signal transducers Stat1 and Stat3 and their novel target Jmjd3 drive the expression of inflammatory genes in microglia.. Journal of Molecular Medicine 2013, 92, 239-54, 10.1007/s00109-013-1090-5.
- Ting Liu; Lingyun Zhang; Donghyun Joo; Shao-Cong Sun; NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy 2017, 2, 17023, 10.1038/sigtrans.2017.23.
- Marion Mussbacher; Manuel Salzmann; Christine Brostjan; Bastian Hoesel; Christian Schoergenhofer; Hannes Datler; Philipp Hohensinner; J. Basílio; Peter Petzelbauer; Alice Assinger; et al.Johannes A. Schmid Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Frontiers in Immunology 2019, 10, 85, 10.3389/fimmu.2019.00085.
- Sergei I. Grivennikov; Michael Karin; Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine & Growth Factor Reviews 2009, 21, 11-9, 10.1016/j.cytogfr.2009.11.005.
- Yihui Fan; Renfang Mao; Jianhua Yang; NF-κB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein & Cell 2013, 4, 176-185, 10.1007/s13238-013-2084-3.
- Oliver Krämer; Daniela Baus; Shirley Knauer; Stefan Stein; Elke Jäger; Roland H. Stauber; Manuel Grez; Edith Pfitzner; Thorsten Heinzel; Acetylation of Stat1 modulates NF-κB activity. Genes & Development 2006, 20, 473-485, 10.1101/gad.364306.
- Linli Yao; Enci Mary Kan; J. Lu; Aijun Hao; S. Thameem Dheen; Charanjit Kaur; Eng-Ang Ling; Toll-like receptor 4 mediates microglial activation and production of inflammatory mediators in neonatal rat brain following hypoxia: role of TLR4 in hypoxic microglia. Journal of Neuroinflammation 2013, 10, 23-23, 10.1186/1742-2094-10-23.
- Min Song; Jingji Jin; Jeong-Eun Lim; Jinghong Kou; Abhinandan Pattanayak; Jamaal Rehman; Hong-Duck Kim; Kazuki Tahara; Robert LaLonde; Ken-Ichiro Fukuchi; et al. TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease. Journal of Neuroinflammation 2011, 8, 92-92, 10.1186/1742-2094-8-92.
- Elvira Akhmetzyanova; Konstantin Kletenkov; Yana Mukhamedshina; Albert Rizvanov; Different Approaches to Modulation of Microglia Phenotypes After Spinal Cord Injury.. Frontiers in Systems Neuroscience 2019, 13, 37, 10.3389/fnsys.2019.00037.
- Kelly Ceyzériat; Lucile Ben Haim; Audrey Denizot; Dylan Pommier; Marco Matos; Océane Guillemaud; Marie-Ange Palomares; Laurene Abjean; Fanny Petit; Pauline Gipchtein; et al.Marie-Claude GaillardMartine GuillermierSueva BernierMylène GaudinGwenaëlle AuréganCharlène JoséphineNathalie DéchampsJulien VeranValentin LanglaisKarine CambonAlexis P BemelmansJan BaijerGilles BonventoMarc DhenainJean-François DeleuzeStéphane H R OlietEmmanuel BrouilletPhilippe HantrayeMaria-Angeles Carrillo-De SauvageRobert OlasoAude PanatierCarole Escartin Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathologica Communications 2018, 6, 104, 10.1186/s40478-018-0606-1.
- Liansheng Zhong; Xue Jiang; Zhihui Zhu; Haiyan Qin; Michael B. Dinkins; Ji-Na Kong; Silvia Leanhart; Rebecca Wang; Ahmed Elsherbini; Erhard Bieberich; et al.Yujie ZhaoGuanghu Wang Lipid transporter Spns2 promotes microglia pro-inflammatory activation in response to amyloid-beta peptide. Glia 2018, 67, 498-511, 10.1002/glia.23558.
- Juan Ji; Juan Wang; Jin Yang; Xi-Peng Wang; Jing-Jing Huang; Teng-Fei Xue; Xiu-Lan Sun; The Intra-nuclear SphK2-S1P Axis Facilitates M1-to-M2 Shift of Microglia via Suppressing HDAC1-Mediated KLF4 Deacetylation.. Frontiers in Immunology 2019, 10, 1241, 10.3389/fimmu.2019.01241.
- Shu Yao; Longjun Li; Xin Sun; Jun Hua; Keqi Zhang; Li Hao; Lixin Liu; Dongyan Shi; Hong Zhou; FTY720 Inhibits MPP+-Induced Microglial Activation by Affecting NLRP3 Inflammasome Activation. Journal of Neuroimmune Pharmacology 2019, 14, 478-492, 10.1007/s11481-019-09843-4.
- Hannah Scheiblich; Anna Schlütter; Douglas T. Golenbock; Eicke Latz; Michael T. Heneka; Pilar Martinez-Martinez; Activation of the NLRP3 inflammasome in microglia: the role of ceramide. Journal of Neurochemistry 2017, 143, 534-550, 10.1111/jnc.14225.
- Michela Campolo; Giovanna Casili; Flavia Biundo; Rosalia Crupi; Marika Cordaro; Salvatore Cuzzocrea; Emanuela Esposito; The Neuroprotective Effect of Dimethyl Fumarate in an MPTP-Mouse Model of Parkinson's Disease: Involvement of Reactive Oxygen Species/Nuclear Factor-κB/Nuclear Transcription Factor Related to NF-E2.. Antioxidants & Redox Signaling 2017, 27, 453-471, 10.1089/ars.2016.6800.
- Marika Cordaro; Giovanna Casili; Irene Paterniti; Salvatore Cuzzocrea; Emanuela Esposito; Fumaric Acid Esters Attenuate Secondary Degeneration after Spinal Cord Injury. Journal of Neurotrauma 2017, 34, 3027-3040, 10.1089/neu.2016.4678.
- Antonio Cuadrado; Sebastian Kügler; Isabel Lastres-Becker; Pharmacological targeting of GSK-3 and NRF2 provides neuroprotection in a preclinical model of tauopathy.. Redox Biology 2017, 14, 522-534, 10.1016/j.redox.2017.10.010.
- Haiyan Peng; Maria Matos; Brian T. Wipke; Moore H. Arnold; Robert Scannevin; Dimethyl fumarate alters microglia phenotype and protects neurons against proinflammatory toxic microenvironments. Journal of Neuroimmunology 2014, 275, 224-44, 10.1016/j.jneuroim.2014.08.602.
- Raffaela Cipriani; Juan Carlos Chara; Alfredo Rodríguez-Antigüedad; Carlos Matute; FTY720 attenuates excitotoxicity and neuroinflammation.. Journal of Neuroinflammation 2015, 12, 86, 10.1186/s12974-015-0308-6.
- Manoj K. Mishra; Janet Wang; Michael B. Keough; Yan Fan; Claudia Silva; Scott Sloka; Liat Hayardeny; Wolfgang Brück; V. Wee Yong; Laquinimod reduces neuroaxonal injury through inhibiting microglial activation. Annals of Clinical and Translational Neurology 2014, 1, 409-422, 10.1002/acn3.67.
- Veit Rothhammer; Jessica E. Kenison; Emily Tjon; Maisa C. Takenaka; Kalil Alves De Lima; Davis M. Borucki; Chun-Cheih Chao; Annabel Wilz; Manon Blain; Luke Healy; et al.Jack AntelFrancisco J Quintana Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proceedings of the National Academy of Sciences 2017, 114, 2012-2017, 10.1073/pnas.1615413114.
- Andrés Miguez; Gerardo García-Díaz Barriga; Veronica Brito; Marco Straccia; Albert Giralt; Silvia Ginés; Josep M. Canals; Jordi Alberch; Gerardo García-Díaz Barriga; Fingolimod (FTY720) enhances hippocampal synaptic plasticity and memory in Huntington's disease by preventing p75NTRup-regulation and astrocyte-mediated inflammation. Human Molecular Genetics 2015, 24, 4958-4970, 10.1093/hmg/ddv218.
- Byung Joo Lee; Jun Young Kim; Hyung-Jung Cho; Donghwi Park; Sphingosine 1-phosphate receptor modulation attenuate mechanical allodynia in mouse model of chronic complex regional pain syndrome by suppressing pathogenic astrocyte activation. Regional Anesthesia & Pain Medicine 2020, 45, 230-238, 10.1136/rapm-2019-100801.
- Emanuela Colombo; Marco Di Dario; Eleonora Capitolo; Linda Chaabane; Jia Newcombe; Gianvito Martino; Cinthia Farina; Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Annals of Neurology 2014, 76, 325-337, 10.1002/ana.24217.
- Yin-Feng Dong; Ruo-Bing Guo; Juan Ji; Lu-Lu Cao; Ling Zhang; Zheng-Zhen Chen; Ji-Ye Huang; Jin Wu; Jun Lu; Xiu-Lan Sun; et al. S1PR3 is essential for phosphorylated fingolimod to protect astrocytes against oxygen‐glucose deprivation‐induced neuroinflammation via inhibiting TLR2/4‐NFκB signalling. Journal of Cellular and Molecular Medicine 2018, 22, 3159-3166, 10.1111/jcmm.13596.
- Saša Trkov Bobnar; Matjaž Stenovec; Katarina Miš; Sergej Pirkmajer; Robert Zorec; Fingolimod Suppresses the Proinflammatory Status of Interferon-γ-Activated Cultured Rat Astrocytes. Molecular Neurobiology 2019, 56, 5971-5986, 10.1007/s12035-019-1481-x.
- Timothy M. Doyle; Zhoumou Chen; MariaConcetta Durante; Daniela Salvemini; Activation of Sphingosine-1-Phosphate Receptor 1 in the Spinal Cord Produces Mechanohypersensitivity Through the Activation of Inflammasome and IL-1β Pathway. The Journal of Pain 2019, 20, 956-964, 10.1016/j.jpain.2019.02.007.
- Nadine Kramann; Lena Menken; Liat Hayardeny; Uwe-Karsten Hanisch; Wolfgang Brück; Laquinimod prevents cuprizone-induced demyelination independent of Toll-like receptor signaling.. Neurology - Neuroimmunology Neuroinflammation 2016, 3, e233, 10.1212/NXI.0000000000000233.
- Wolfgang Brück; Ramona Pförtner; Trinh Pham; Jingya Zhang; Liat Hayardeny; Victor Piryatinsky; Uwe-Karsten Hanisch; Tommy Regen; Denise Van Rossum; Lars Brakelmann; et al.Karin HagemeierTanja KuhlmannChristine StadelmannGareth R. JohnNadine KramannChristiane Wegner Reduced astrocytic NF-κB activation by laquinimod protects from cuprizone-induced demyelination. Acta Neuropathologica 2012, 124, 411-24, 10.1007/s00401-012-1009-1.
- Laura Weinstock; Amanda M Furness; Shawn S Herron; Sierra S Smith; Sitara B. Sankar; Samantha G DeRosa; Dadi Gao; Molly E Mepyans; Anna Scotto Rosato; Diego Luis Medina; et al.Ayelet VardiNatalia S FerreiraSoo Min ChoAnthony H. FutermanSusan A. SlaugenhauptLevi B. WoodYulia Grishchuk Fingolimod phosphate inhibits astrocyte inflammatory activity in mucolipidosis IV. Human Molecular Genetics 2018, 27, 2725-2738, 10.1093/hmg/ddy182.
- Qiao Ling Cui; Jun Fang; Timothy E. Kennedy; Guillermina Almazan; Jack P. Antel; Role of p38MAPK in S1P receptor-mediated differentiation of human oligodendrocyte progenitors. Glia 2014, 62, 1361-1375, 10.1002/glia.22688.
- Maribel Osinde; Florian Mullershausen; Kumlesh K. Dev; Phosphorylated FTY720 stimulates ERK phosphorylation in astrocytes via S1P receptors. Neuropharmacology 2007, 52, 1210-1218, 10.1016/j.neuropharm.2006.11.010.
- Peng Li; Gang Zhao; Yan Ding; Tianyi Wang; Jerry Flores; Umut Ocak; Pei Wu; Tongyu Zhang; Jun Mo; John H. Zhang; et al.Jiping Tang Rh-IFN-α attenuates neuroinflammation and improves neurological function by inhibiting NF-κB through JAK1-STAT1/TRAF3 pathway in an experimental GMH rat model.. Brain, Behavior, and Immunity 2019, 79, 174-185, 10.1016/j.bbi.2019.01.028.
- Ethan R. Roy; Baiping Wang; Ying-Wooi Wan; Gabriel S. Chiu; Allysa L. Cole; Zhuoran Yin; Nicholas E. Propson; Yin Xu; Joanna L. Jankowsky; Zhandong Liu; et al.Virginia M.-Y. LeeJohn Q. TrojanowskiStephen D. GinsbergOleg ButovskyHui ZhengWei Cao Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. Journal of Clinical Investigation 2020, 130, 1912-1930, 10.1172/jci133737.
- Tanusree Sen; Pampa Saha; Rajaneesh Gupta; Lesley M. Foley; Tong Jiang; Olena S. Abakumova; Kevin T. Hitchens; Nilkantha Sen; Aberrant ER Stress Induced Neuronal-IFNβ Elicits White Matter Injury Due to Microglial Activation and T-Cell Infiltration after TBI. The Journal of Neuroscience 2019, 40, 424-446, 10.1523/jneurosci.0718-19.2019.
- Kazuo Nakamichi; Megumi Saiki; Hiroshi Kitani; Yuki Kuboyama; Kinjiro Morimoto; Mutsuyo Takayama-Ito; Ichiro Kurane; Suppressive effect of simvastatin on interferon-β-induced expression of CC chemokine ligand 5 in microglia. Neuroscience Letters 2006, 407, 205-210, 10.1016/j.neulet.2006.08.044.
- Wen Li; Barney Viengkhou; Gareth Denyer; Phillip West; Iain L. Campbell; Markus J. Hofer; Microglia have a more extensive and divergent response to interferon-α compared with astrocytes. Glia 2018, 66, 2058-2078, 10.1002/glia.23460.
- Veit Rothhammer; Ivan D. Mascanfroni; Lukas Bunse; Maisa C. Takenaka; Jessica E. Kenison; Lior Mayo; Chun-Cheih Chao; Bonny Patel; Raymond Yan; Manon Blain; et al.Jorge I. AlvarezHania KébirNiroshana AnandasabapathyGuillermo IzquierdoSteffen JungNikolaus ObholzerNathalie PochetClary ClishMarco PrinzAlexandre PratJack AntelFrancisco J Quintana Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nature Medicine 2016, 22, 586-597, 10.1038/nm.4106.
- Olga Barca; Pablo Devesa-Peleteiro; Marcos Seoane; Rosa Señarís; Victor Arce; Bimodal effect of interferon-β on astrocyte proliferation and survival: Importance of nuclear factor-κB. Journal of Neuroimmunology 2010, 226, 73-80, 10.1016/j.jneuroim.2010.05.036.
- Brad A. Friedman; Karpagam Srinivasan; Gai Ayalon; William J. Meilandt; Han Lin; Melanie A. Huntley; Yi Cao; Seung-Hye Lee; Patrick C.G. Haddick; Hai Ngu; et al.Zora ModrusanJessica L. LarsonJoshua S. KaminkerMarcel Van Der BrugDavid V. Hansen Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Reports 2018, 22, 832-847, 10.1016/j.celrep.2017.12.066.