Viruses have evolved different strategies to hijack subcellular organelles during their life cycle to produce robust infectious progeny. Successful viral reproduction requires the precise assembly of progeny virions from viral genomes, structural proteins, and membrane components. Such spatial and temporal separation of assembly reactions depends on accurate coordination among intracellular compartmentalization in multiple organelles. Virus trigger the rearrangement and morphology remodeling of intracellular organelles, including the quality control of intracellular organelles, the hijacking of the modified organelle membranes, morphology remodeling for viral replication, and degradation of intracellular organelles by virus-triggered selective autophagy.
- virus
- intracellular organelles
- rearrangement
- remodeling
1.Rearrangement of Introduintracellular organelles during viral infections
To maximize their viral replication and evade host antiviral responses, viruses have evolved a plethora of strategies to hijack cellular organelles [1][2][3]. Each step of viral replication is closely accompanied by the rearrangement of intracellular organelles.
2. Remodeling of the mitochondria for viral replication
The mitochondria are highly dynamic organelles and form interconnected tubular networks, undergoing a balance between fusion and fission in response to intracellular and/or extracellular stresses [4] (Figures 1A and 1B). Mitochondrial fusion involves two sets of key GTPase proteins in mammals: the mitofusin GTPases (Mfns) (Mfn1 and Mfn2) of the outer mitochondrial membrane (OMM) and optic atrophy 1 (OPA1) of the inner mitochondrial membrane (IMM) [5][6][7][8][9]. The Mfns mediate OMM fusion and cristae integrity [10]. However, the OPA1 mediates IMM fusion and cristae integrity by regulating of the mRNA splicing forms, membrane potential, and the adenosine triphosphate (ATP)-dependent diverse cellular proteases [6][7][8]. Subsequently, OMM fusions are followed by IMM fusion processes, resulting in the concomitant mixing of the mitochondrial contents and merging of two individual mitochondria. In a previous study, Cipolat, S et al. identified that OPA1 specific functional cross-talk with Mfn1 rather than Mfn2 is involved in the mitochondrial fusion of OMM [11]. Mitochondrial fission is a complex process which includes two distinct steps: an initial constriction of mitochondrial membranes and membrane scission. The initial constriction step narrows the mitochondrial tube diameter at the ER-mitochondria intersection zones where ER tubules wrap around the OMM. Manor, U. et al. suggested that actin-nucleating protein spire 1C localizes to the mitochondria, and directly links the mitochondria to the actin cytoskeleton and the ER, and finally promotes actin polymerization at the ER-mitochondria intersections [12]. The membrane scission of the mitochondria is primarily regulated by dynamic relative GTPase protein (DRP-1) [13]. The mitochondrial localization of DRP-1 is a cytosolic factor promoting mitochondrial fission, which powers the constriction and division of the mitochondria primarily through post-translational modification (e.g., phosphorylation) (reviewed by Lee. H et al. [14]). Recent studies have reported that the recruitment of DRP-1 in mammalian cells requires several accessory proteins, such as the mitochondrial fission protein 1 (Fis-1) and mitochondrial fission factor (Mff) [15]. Although such proteins are proposed to constitute the fission complex of the mitochondria, mediating mitochondrial fission using this complex has remained unclear.
Viruses have evolved several strategies to remodel the mitochondria for viral replication and assembly, including spatial distribution, morphology remodeling, and metabolism reprogramming. To maximize the effectiveness of DNA replication, African swine fever virus (ASFV) infection recruits the mitochondria around the viral factories, associated with the morphology change and accumulation of the mitochondria. It was speculated that the translation and ATP synthesis are coupled and compartmentalized around viral factories to promote virus replication [16] (Figure 1C). Normal mitochondria are dynamic organelles, and form interconnected tubular networks [4][17] (Figure 1A). The cristae remodeling of the IMM determines the assembly and stability of respiratory chain supercomplexes and respiratory efficiency [18]. In general, the mitochondrial elongation process is associated with the dimerization and activation of the ATPase function to produce additional energy [17][19]. NDV induces the hyper-fusion of the mitochondria in infected A549 cells (unpublished data), which is similar to the characteristic of Dengue virus (DENV) [20] and severe acute respiratory syndrome-coronavirus (SARS-CoVs) [21]. Notably, except for vaccinia virus (VV) [22], most viruses exploit aerobic glycolysis of the mitochondria for the production of viral progeny [2].
Moreover, viral infections may increase the inter-organellar interactions of the mitochondria with other organelles for replication. Rubella virus (RUBV) [23] and Bunyamwera virus (BUNV) [24] infections increase the membrane interactions among mitochondria, ER, and Golgi (Figure 1C), which is consistent with that of the ER-mitochondria contract that serves as a platform for inter-organellar communication [25].
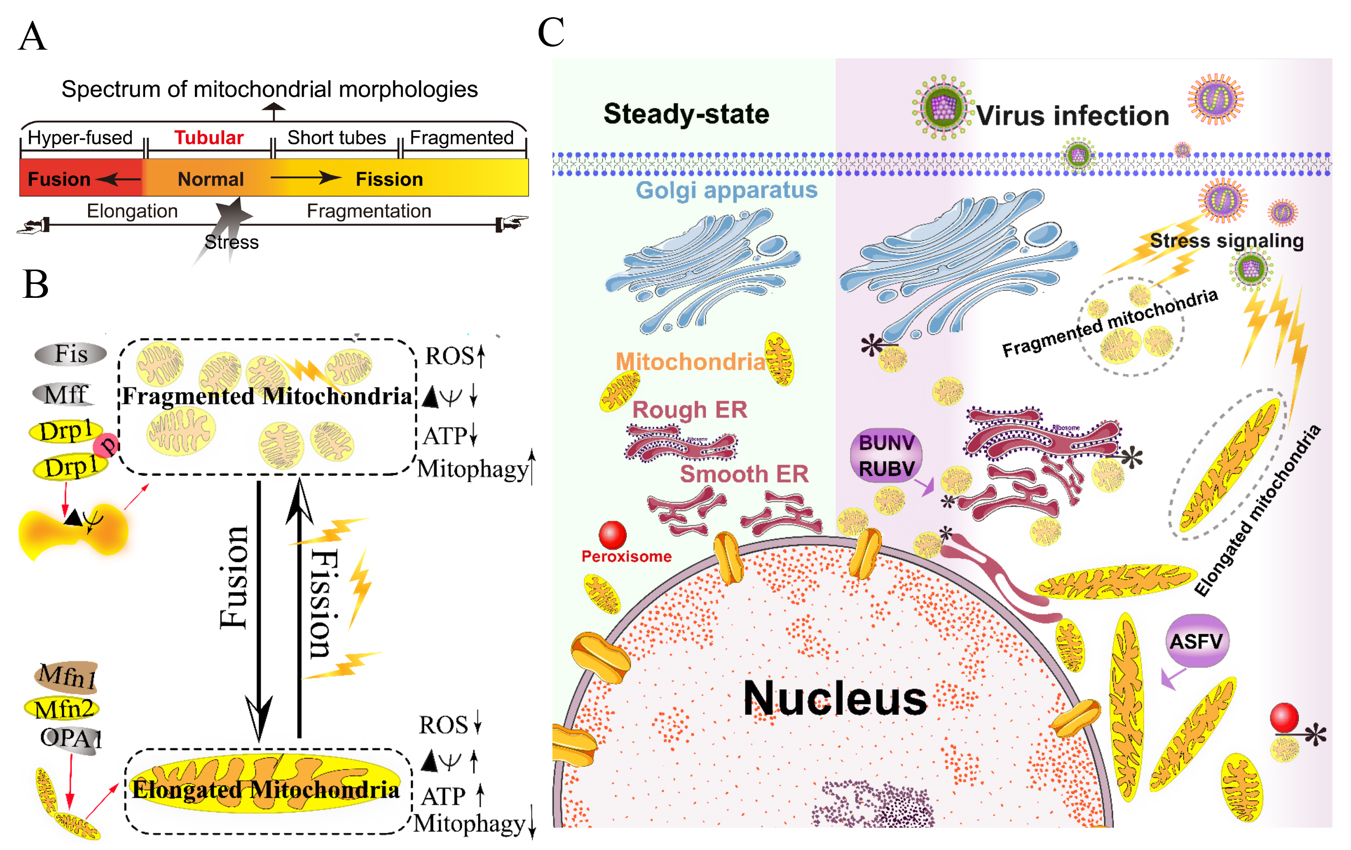
Figure 1. Morphology remodeling and spatial redistribution of mitochondria triggered by virus infections. (A) The morphological diagram of the mitochondria. Mitochondria form a dynamic network pool, which constantly undergoes rearrangement and turnover. The equilibrium regulation of mitochondrial fusion–fission is essential to maintain the integrity of mitochondria [26]. The morphology of mitochondria was divided into hyper-fused (elongated), tubular (normal), short tubes, and fragmented [17]. (B) Regulation of mitochondrial fusion and fission. Mitochondrial fusion is mediated by mitofusin GTPases MFN1 and MFN2 at the outer mitochondrial membrane (OMM), and OPA1 at the inner mitochondrial membrane (IMM). Mitochondrial fission is driven by the fission machinery complex, which consists of DRP-1, Fis1, and MFF. Mitochondrial hyper-fusion is a pro-survival type, which can increase the ATP production and membrane potential (Δψm), and decrease reactive oxygen species (ROS) and mitophagy [17][26]. (C) Proposed model for the nuclear aggregation of mitochondria and the possible interplay among intracellular organelles in response to virus infections. * symbol indicates the possible interaction site. Representative virus that increases the interactions among intracellular organelles is shown with purple rectangle. African swine fever virus, ASFV; Rubella virus, RUBV; Bunyamwera virus, BUNV.
To date, several reports have argued the role of Mfns in innate immunity [27][28][29]. The interaction of Mfns with the adaptor mitochondrial antiviral signaling protein (MAVS) (also called IPS-1, Cardif, or VISA) at the mitochondrial associated membrane (MAM) leads the initiation of the IFN signaling pathway [30][31]. Meanwhile, MAVS was also reported to interact with MFN2, which leads to the inhibition of inflammatory cytokine production, suggesting the MAM plays a complex role in the regulation of innate immunity [28] (detailed in review [31]). Castanier, C et al. also identified the cross-modulation relationship between mitochondrial dynamic and retinoic acid-inducible gene I protein (RIG-I) like receptor (RLR) signaling activation [27]. Certain viruses, such as influenza A virus (IAV) [32], measles virus (MV) [33], hepatitis B virus (HBV) [34][35][36][37], and hepatitis C virus (HCV) [34], induce selective autophagy to degrade fragmented mitochondria and evade innate immunity. Meanwhile, the non-structural (NS) protein 4B of DENV induces mitochondrial elongation via inactivation of DRP-1 and dampens the activation of RLR signal pathway to promote replication [20]. Similarly, the open reading frame-9b (ORF-9b) encoded by SARS-CoVs causes mitochondrial elongation via triggering DRP-1 degradation, and inhibits RLR signaling [21].
Collectively, viruses have evolved several strategies to hijack the mitochondria for viral genome replication and assembly, including the remodeling of mitochondrial morphology and distribution, the regulation of the fusion–fission machinery complex, and the synthesis of ATP production.
2.1. Rearrangement of ER and unfolded protein response (UPR) during viral infection
The ER, a single continuous membrane, consists of two primary structural subdomains: the nuclear envelope and the peripheral ER (a polygonal network) [38]. The nuclear envelope of ER consists of two flat membrane bilayers; the peripheral ER is composed of membrane cisternae and dynamic interconnected tubules [38][39]. The ER is the largest intracellular endomembrane system and has multiple complex functions, including Ca2+ storage, fatty acid synthesis, ion homeostasis, and, in particular, the quality control of newly synthesized proteins [40]. The accumulation of misfolded or unfolded proteins in the ER lumen is known as ER stress [41]. UPR and ER-associated degradation (ERAD) signaling are central to maintain the quality control of the ER [41][42]. The UPR is a signaling cascade aimed at eliminating misfolding proteins and increasing folding capacity in lumen [41]. The protein-folding conditions in the ER lumen is primarily sensed by three integrated signaling transducers: activating transcription factor 6 (ATF6) [43], double-stranded RNA-activated protein kinase-like kinase (PERK), and inositol requiring enzyme 1α (IRE1α) [25][44] (Figure 2). Each branch uses a distinct mechanism to drive the transcription of UPR signal transduction, such as ATF6 by regulated proteolysis, PERK by translational control, and IRE1 by non-conventional mRNA splicing [44]. By contrast, ERAD recognizes misfolded proteins and retro-translocates such proteins into the cytoplasm for degradation by the ubiquitin-proteasome-dependent ERAD and the autophagy-lysosome dependent ERAD [42][45].
A series of studies has reported that viral infections reshape the morphology and membrane remodeling of ER [3][38], and exploit various strategies to hijack the three branches of UPR for viral replication (Figure 2 and Table 1). The possible explanations were summarized as follows: First, the large malleable surface area of ER is used as a physical scaffold to protect viral RNA from degradation by cellular mRNA decay machinery [40][46]. RNA viruses have evolved several strategies to avoid the cellular mRNA decay machinery [46]. Second, viruses, particularly most RNA viruses, remodel the ER membrane to form a variety of structures for infectious progeny [47], including single-membrane spherule vesicles, double-membrane vesicles, convoluted membranes, and single-membrane sheets in the ER lumen [38]. Tenorio, R et al. identified that δNS and μNS of reovirus caused tubulation and fragmentation of the ER, respectively, to re-build replication sites [48], indicating that viral proteins play different roles in the rearrangement of ER membranes. Similarly, the NS4A of DENV induces the membrane arrangement of ER lumen in a 2K-regulated manner [49]. Third, viruses recruit the ER membranes into the replication and assembly compartments. The viral cytoplasmic replication site of VV [50][51], equine arteritis virus (EAV) [52], and polivirus (PV) [53] is derived directly from the ER membrane. Meanwhile, ASFV structural protein p54 plays an important role in the recruitment and transformation of the ER membranes into the envelope precursors [54]. Fourth, viruses increase the capacity and spatial rearrangement to increase ER biogenesis, including membrane protein synthesis, fatty acid change, and Ca2+ storage [40]. For enveloped viruses, the key molecular chaperone of ER, including Bip/GRP78 and calnexin/calreticulin, assists the folding of the extracellular domains of viral membrane glycoproteins, such as GP2a of PRRSV [55], and hemagglutinin-neuraminidase (HN) and fusion (F) proteins of NDV [56], when they translocate into the lumen of the ER. Meanwhile, the reprograming of ER biogenesis, such as Ca2+ storage, is required for viral replication, including HCV [57] and ASFV [58]. Fifth, viruses co-opt or subvert the ERAD processes to re-establish ER homeostasis, which actively exports the malformed proteins from the ER for degradation. Human cytomegalovirus (HCMV) [59] and IAV [60] exploit the ERAD pathway to benefit viral replication. Finally, the membrane remodeling of ER may suppress the activation of host immunity. Upon viral infections, particularly DNA viruses, stimulator of interferon genes (STING), an activated ER adaptor of the cyclic GMP-AMP synthase (cGAS)-STING signaling pathway, translocates from the ER to the ER-Golgi-intermediate compartment (ERGIC) and the Golgi apparatus, and then activates downstream molecules [61][62][63]. Therefore, we speculate that the morphology remodeling and membrane modification of ER induced by viruses may be involved in the regulation of STING trafficking, EARD degradation, and post-translational modification, and eventually evade the activation of cGAS-STING pathway.
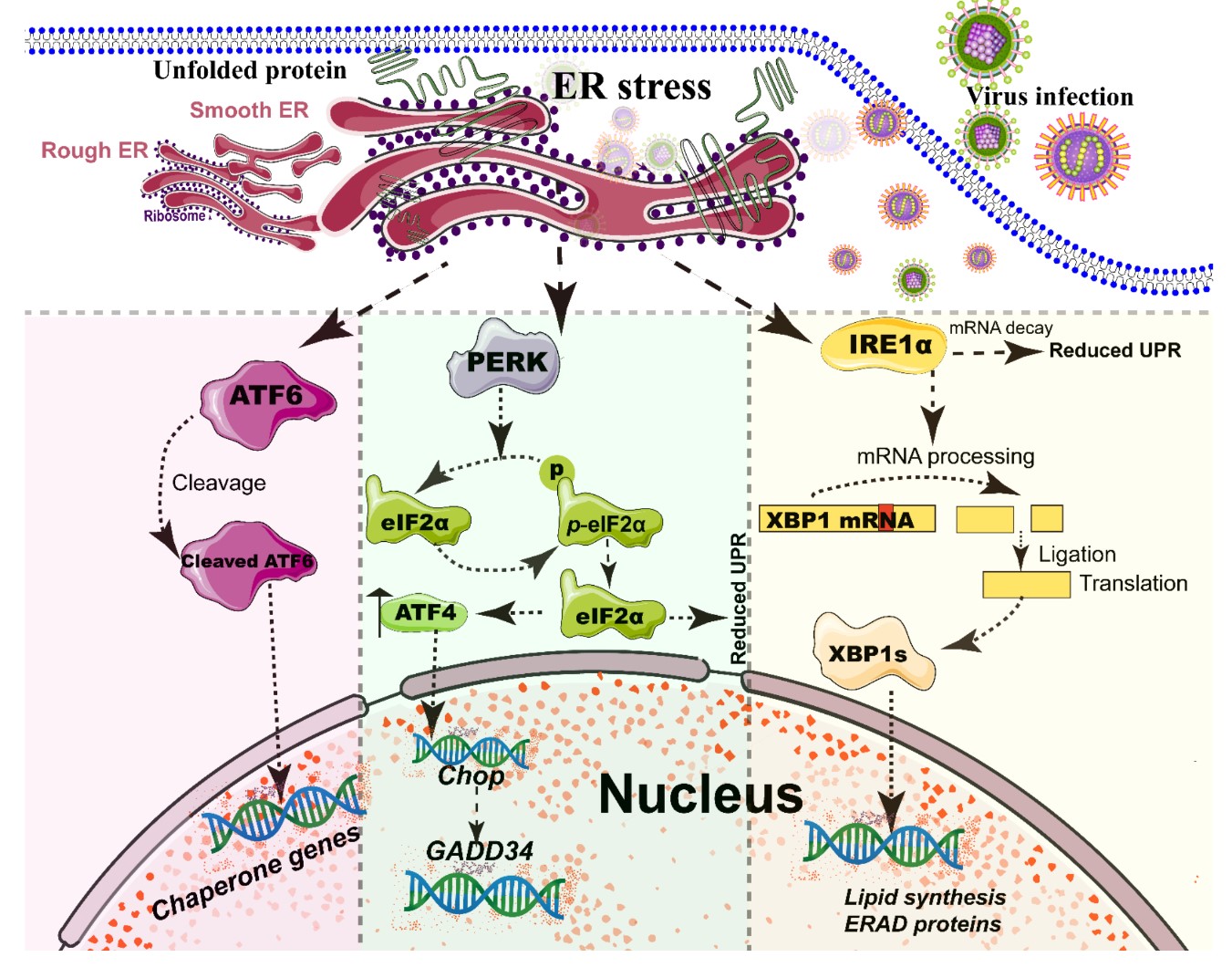
Figure 2. Simplified diagram of the core element of the three unfolded protein response (UPR) signaling branches of the endoplasmic reticulum (ER). During different viral infections, the ER stress activates the three stress sensor proteins: IRE1α, ATF6, and PERK (Detailed in reviews [44][64]). Each sensor uses a distinct mechanism of signal transduction to drive the transcription of UPR target genes and eventually work as feedback loops to mitigate the ER stress [44][64]. Upon ER stresses, ATF6, a transcriptional factor, translocate into the Golgi compartment where it is cleaved by the site (1/2) protease. The N-terminal cytosolic domain of cleaved ATF6 is released into cytosol and then translocated into the nucleus where it binds to ER stress-response elements to activate target genes, including XBP-1 and C/EBP-homologous protein (CHOP) [43]. The activation of PERK inhibits general protein translation by the phosphorylation of eIF2α, enabling dedicated translation of transcripts, including ATF4, a key transducer. The IRE1 branch is regulated by non-conventional mRNA splicing [44][64]. Subsequently, the activated IRE1 processes XBP1 mRNA to generate the spliced form of XBP1 protein (XBP1s), which participates in IRE1α-mediated UPR pathway in response to ER stresses [44][64]. Eventually, the activation of the cleaved ATF6 (N-ATF6), ATF4, and XBP1 transcription factors increases the protein-folding capacity in the ER lumen. Meanwhile, IRE1 and PERK sensors also decrease the load of proteins entering the ER [44][64].
Table 1. Viruses activate and exploit the UPR branch of ER for viral replication.
|
Viruses |
Family |
Genome Structure |
Virion Structure |
Viral Protein |
ATF6 |
PERK |
IRE1α |
Ref |
|
PRRSV |
Arteriviridae |
Linear, ssRNA(+) |
Enveloped; Spherical |
? |
× |
√ |
√ |
[55] |
|
IBV |
Coronaviridae |
Linear, ssRNA(+) |
Enveloped; Spherical |
? |
× |
× |
√ |
[63] |
|
DENV |
Flaviridae |
Linear, ssRNA(+) |
Enveloped; Spherical |
? |
× |
√ |
√ |
[65] |
|
JEV |
Flaviridae |
Linear, ssRNA(+) |
Enveloped; Spherical |
NS4B |
√ |
√ |
√ |
|
|
TBEV |
Flaviridae |
Linear, ssRNA(+) |
Enveloped; Rounded |
? |
√ |
× |
√ |
[69] |
|
HCV |
Flaviridae |
Linear, ssRNA(+) |
Enveloped; Spherical |
? |
√ |
× |
× |
[70] |
|
IAV |
Orthomyxoviridae |
Segmented, ssRNA (-) |
Enveloped; Rounded |
? |
× |
|
√ |
[71] |
|
NDV |
Paramyxoviridae |
Linear, ssRNA(-) |
Enveloped; Spherical |
F and HN |
√ |
√ |
√ |
|
|
MCMV |
Herpesviridae |
Linear, ds DNA |
Enveloped; Spherical |
? |
? |
√ |
× |
|
|
HSV-1 |
Herpesviridae |
Linear, dsDNA |
Enveloped; Spherical |
UL41/ICP0/γ134.5 |
√ |
√ |
× |
|
|
HBV |
Hepadnaviridae |
Circular dsDNA |
Enveloped; Spherical |
? |
√ |
× |
√ |
[79] |
During different viral infections, the ER stress activates the three stress sensor proteins: IRE1α, ATF6, and PERK (review in the references [44][64]). The following abbreviations are used in this table: murine cytomegalovirus, MCMV; avian coronavirus infectious bronchitis virus, IBV; porcine reproductive and respiratory syndrome virus, PRRSV ; hepatitis C virus, HCV; hepatitis B virus, HBV; influenza A virus, IAV; human herpes simplex virus-1, HSV-1; dengue virus, DENV; tick-borne encephalitis virus, TBEV; Japanese encephalitis virus, JEV; Newcastle disease virus, NDV. The symbols √, ×, and ? indicate activation, inhibition, and unknown, respectively.
2.2. Rearrangement of peroxisome for infectious progeny
The peroxisomes are single membrane-bounded organelles that function in numerous metabolic pathways, including β-oxidation of long-chain fatty acids, detoxification of hydrogen peroxide, and synthesis of ether phospholipids and bile acids [80][81][81, 82]. Notably, the mitochondria and peroxisomes share common functions in the β-oxidation of fatty acids and the reduction of damaging peroxides. Proliferation of peroxisome is largely mediated by growth and division. Peroxisomal division in mammalian cells comprises multiple processes, including membrane deformation, elongation, constriction, and fission [82]. With the exception of peroxin (PEX)11, the peroxisomes and mitochondria share common fission machinery, including DRP-1, Mff, and Fis1 [83][84]. The fission machinery of peroxisome is orchestrated by PEX-11β and mitochondrial fission factors [82]. Mitochondrial-derived vesicles (MDVs) are involved in the transportation of mitochondrial-anchored protein ligase (MAPL), a mitochondrial outer membrane, to peroxisomes [85]. The retromer complex containing vacuolar protein sorting (Vps) 5, Vps 26, and Vps 29, a known component of vesicle transport from the endosome to the Golgi apparatus, also regulates the transport of MAPL as a binding partner from the mitochondria to peroxisomes [86].
Viruses regulate the morphology and biogenesis of peroxisomes to promote progeny replication [1]. For instance, the C-terminal of the rotavirus VP4 protein is directly located in peroxisomes via its conserved peroxisomal targeting signal [87]. Meanwhile, viruses have exploited the myristoyl-CoA produced by peroxisome biogenesis for the N-myristoylation of viral proteins [1], such as ASFV [88], indicating that peroxisomal lipid metabolism contributes to viral replication. Another typical example is the tomato bushy stunt virus (TBSV), a member of the Tombusviridae family, which infects a variety of plant species. McCartney, A.W et al. reported that TBSV induced the rearrangement of peroxisomes and caused vesiculation of the peroxisomal membrane, where it provided a scaffold for virus anchoring and server as the sites of viral RNA synthesis [89]. In the absence of peroxisomes, TBSV also exploits the surface of the ER membrane as a viral factory for viral replication and assembly [90]. It is suggestive of the remarkable flexibility of the virus to use host membranes for replication.
2.3. Hijacking of Golgi apparatus for infectious progeny
The Golgi apparatus is a highly dynamic organelle whose function primarily includes saccule formation and utilization of such saccules in vesicle formation at the opposite face for delivery [91]. The normal cellular secretory pathway, bidirectional transport between the ER and Golgi apparatus, is mediated by tubulovesicular transport containers that depend on two coat protein complexes, COP-I and COP-II. The COP-II establishes a membrane flow from the ER to the Golgi complex [92]. However, the COP-I coat acts in retrograde transport from the Golgi back to the ER [93].
In general, certain viruses utilize the cellular trafficking and secretory pathway of the ER-Golgi transport system to replicate/release their progeny [3]. PV utilizes the components of the ADP-ribosylation factor (Arf) family of small GTPases [94] and cellular COP-II proteins [95] to form membrane-bound replication complex for viral replication, indicating that PV hijacks the components of the cellular secretory pathway for replication. As shown in Table 2, metonaviridae [23] and peribunyaviridae [24] hijack the Golgi complex to re-construct it as a viral factory for viral replication. For example, RUBV [23] and BUNV [24] infections modify cell structural morphology and remodel the Golgi complex as a viral factory during the entire life cycle. Furthermore, host secretory signaling is also crucial for innate and acquired immune responses, such as the exportation of proinflammatory and antiviral cytokines. Nearly 25 years ago, Doedens, J.R et al. reported that the 2B and 3A proteins of PV inhibited cellular protein secretion by directly blocking the transportation from the ER to the Golgi apparatus [96], indicating that the functional secretory protein is not indispensable for viral RNA replication. Mechanistically, Dodd, D.A et al. identified that the inhibition of 3A protein of PV on the ER to Golgi limited the antiviral cytokine secretion of native immune response and inflammation, including interleukin-6, interleukin-8, and β-interferon [97]. Deitz, S.B et al. also identified that PV 3A protein reduced the presentation of new antigens on the cell surface [98]. Considering that the ER adaptor STING was also located on the Golgi and ERGIC [60], we hypothesized that the membrane remodeling and modification of Golgi induced by viruses might also be involved in the regulation of cGAS-STING pathways (Figure 3). Collectively, all these data suggest that enteroviruses, such as PV and CVB, have evolved a series of strategies to hijack cellular trafficking and secretion for viral replication.
Table 2. Intracellular compartment sites for viral replication and assembly.
|
Family |
Viruses |
Genome Structure |
Virion Structure |
Replication Site |
Ref |
Assembly Site |
Ref |
||
|
Asfarviridae |
ASFV |
Liner, dsDNA |
Enveloped, spherical |
Nuclear and cytoplasmic |
ER |
||||
|
Poxviridae |
VV |
Liner, dsDNA |
Enveloped, brick-shaped |
cytoplasmic |
[101] |
ERGIC, ER |
|||
|
Arteriviridae/Arteriviruses |
EAV, PRRSV |
Liner, ssRNA (+) |
Enveloped, spherical |
Cytoplasmic double membranes |
ER |
[52] |
|||
|
Coronaviridae/Coronavirus |
SARS/MHV |
Liner, ssRNA (+) |
Enveloped, spherical |
Cytoplasmic double membranes |
[103] |
ERGIC, Golgi, ER |
|||
|
Flaviviridae/ Hepacivirus |
HCV |
Linear, ssRNA(+) |
Enveloped, spherical |
Cytoplasmic |
[106] |
Autophagosome |
|||
|
Metonaviridae/Rubivirus |
RUBV |
Liner, ssRNA (+) |
Enveloped, spherical isometric capsid? |
Cytoplasmic |
[23] |
Golgi, Lysosome |
|||
|
Nodaviridae |
FHV |
Linear, ssRNA(+) |
Non-envelop, icosahedral |
Cytoplasmic |
OMM |
||||
|
Togaviridae/Alphavirus |
SFV |
Liner, ssRNA (+) |
Enveloped, spherical and icosahedral |
Cytoplasmic |
Endosome /Lysosome |
||||
|
Tombusviridae |
TBSV |
Linear, ssRNA(+) |
Non-envelop, icosahedral |
Cytoplasmic |
[90] |
Peroxisome, ER |
|||
|
Picornaviridae/Enterovirus |
CV/PV |
Linear, ssRNA(+) |
Non-envelop, spherical |
Cytoplasmic |
[115] |
ER |
|||
|
Orthomyxoviridae |
IAV |
Segmented, ssRNA (-) |
Enveloped; Rounded |
Nuclear |
[116] |
ER, Golgi |
[116] |
||
|
Peribunyaviridae/Bunyavirus |
BUNV |
Segmented, ssRNA (-) |
Enveloped, spherical |
cytoplasmic |
[24] |
Golgi |
|||
|
Retroviridae/Lentivirus |
HIV |
Linear, ssRNA(+) |
Enveloped, Spherical |
Nuclear |
[118] |
ER or Golgi |
[118] |
||
The following abbreviations are used in this table: African swine fever virus, ASFV; rubella virus, RUBV; Bunyamwera virus, BUNV; semliki forest virus, SFV; Flock house virus, FHV; tomato bushy stunt virus, TBSV; severe acute respiratory syndrome, SARS; murine hepatitis virus, MHV; porcine reproductive and respiratory syndrome virus, PRRSV ; equine arteritis virus, EAV; influenza A virus, IAV; human immune deficiency, HIV; vaccinia virus, VV; poliovirus, PV; coxsackieviruses, CV. reticulum-Golgi intermediate compartment, ERGIC; outer mitochondrial membranes, OMM.
2.4. Role of the lysosome and endosome in viral infections
The lysosome, an acidic and membrane-bound organelle, acts as a cellular recycling center and is filled with a number of hydrolases [119]. The lysosome-based degradation processes are subject to reciprocal regulation [120]. Lysosomes degrade unwanted materials which are delivered either from outside via the endocytic pathway or from inside via the autophagic pathway [120][121]. For viral replication and assembly, certain viruses, including Alphaviruses [113], such as semliki forest virus (SFV) [112], exploit the membrane surface of the endosome and lysosome as a viral factory. Similarly, RUBV also can modify the membrane of lysosomes for a viral factory [109]. Meanwhile, the Toll-like receptors (TLR), such as TLR 3/7/9, are located on the endosome, indicating that the endosome also plays an important role in innate immunity [61]. Therefore, we speculate that another possible strategy is that viruses, particularly DNA viruses, evade the TLR-mediated activation of the NF-κB and transcription of proinflammatory cytokines. HBV infection suppresses TLR-9 expression and prevents TLR9 promoter activity in human primary B cells [122]. Interestingly, DENV, a positive-stand RNA virus, activates the TLR9 by inducing mtDNA release in human dendritic cells [123]. Additionally, the endosomal-lysosomal sorting system is a complex and dynamic vesicular sorting signaling, which is fundamental to maintain homeostasis [124]. Viruses, particularly enveloped viruses such as HIV [118], have evolved several strategies to hijack the endosomal sorting complex required for the transport (ESCRT) complex to facilitate viral budding. Collectively, all these data indicate that different viruses utilize different strategies to hijack the endosome/lysosome for viral progeny.
References
- Paul B. Lazarow; Viruses exploiting peroxisomes. Current Opinion in Microbiology 2011, 14, 458-469, 10.1016/j.mib.2011.07.009.
- Erica L. Sanchez; Michael Lagunoff; Viral activation of cellular metabolism. Virology 2015, 479, 609-618, 10.1016/j.virol.2015.02.038.
- Makeda Robinson; Stanford Schor; Rina Barouch-Bentov; Shirit Einav; Viral journeys on the intracellular highways. Cellular and Molecular Life Sciences 2018, 75, 3693-3714, 10.1007/s00018-018-2882-0.
- Gilad Twig; Brigham Hyde; Orian S Shirihai; Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view.. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2008, 1777, 1092-7, 10.1016/j.bbabio.2008.05.001.
- Hsiuchen Chen; Scott A. Detmer; Andrew J. Ewald; Erik E. Griffin; Scott E. Fraser; David Chan; Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. Journal of Cell Biology 2003, 160, 189-200, 10.1083/jcb.200211046.
- Zhiyin Song; Hsiuchen Chen; Maja Fiket; Christiane Alexander; David Chan; OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. Journal of Cell Biology 2007, 178, 749-755, 10.1083/jcb.200704110.
- Sarah Ehses; Ines Raschke; Giuseppe Mancuso; Andrea Bernacchia; Stefan Geimer; Daniel Tondera; Jean-Claude Martinou; Benedikt Westermann; Elena I Rugarli; Thomas Langer; et al. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. Journal of Cell Biology 2009, 187, 1023-1036, 10.1083/jcb.200906084.
- Brian Head; Lorena Griparic; Mandana Amiri; Shilpa Gandre-Babbe; Alexander M. Van Der Bliek; Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. Journal of Cell Biology 2009, 187, 959-966, 10.1083/jcb.200906083.
- Aurélien Olichon; Laurent Baricault; Nicole Gas; Emmanuelle Guillou; A. Valette; Pascale Belenguer; Guy Lenaers; Loss of OPA1 Perturbates the Mitochondrial Inner Membrane Structure and Integrity, Leading to Cytochrome c Release and Apoptosis. Journal of Biological Chemistry 2003, 278, 7743-7746, 10.1074/jbc.c200677200.
- A Santel; M T Fuller; Control of mitochondrial morphology by a human mitofusin. Journal of Cell Science 2001, 114, 867-874.
- Sara Cipolat; Olga Martins De Brito; Barbara Dal Zilio; Luca Scorrano; OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proceedings of the National Academy of Sciences 2004, 101, 15927-15932, 10.1073/pnas.0407043101.
- Uri Manor; Sadie Bartholomew; Gonen Golani; Eric Christenson; Michael Kozlov; Henry N. Higgs; James Spudich; Jennifer Lippincott-Schwartz; A mitochondria-anchored isoform of the actin-nucleating spire protein regulates mitochondrial division. eLife 2015, 4, -, 10.7554/eLife.08828.
- Elena Smirnova; Lorena Griparic; Dixie-Lee Shurland; Alexander M. Van Der Bliek; Dynamin-related Protein Drp1 Is Required for Mitochondrial Division in Mammalian Cells. Molecular Biology of the Cell 2001, 12, 2245-2256.
- Hakjoo Lee; Yisang Yoon; Mitochondrial fission: regulation and ER connection. Molecules and Cells 2014, 37, 89-94, 10.14348/molcells.2014.2329.
- Hidenori Otera; Chunxin Wang; Megan M. Cleland; Kiyoko Setoguchi; Sadaki Yokota; Richard J. Youle; K Mihara; Mff is an essential factor for mitochondrial recruitment of Drp1 during mitochondrial fission in mammalian cells. Journal of Cell Biology 2010, 191, 1141-1158, 10.1083/jcb.201007152.
- Alfredo Castelló; Ana Quintas; Elena G. Sánchez; Prado Sabina; Marisa Nogal; Luis Carrasco; Yolanda Revilla; Regulation of Host Translational Machinery by African Swine Fever Virus. PLOS Pathogens 2009, 5, e1000562, 10.1371/journal.ppat.1000562.
- Timothy Wai; Thomas Langer; Mitochondrial Dynamics and Metabolic Regulation. Trends in Endocrinology & Metabolism 2016, 27, 105-117, 10.1016/j.tem.2015.12.001.
- Sara Cogliati; Christian Frezza; María Eugenia Soriano; Tatiana Varanita; Rubén Quintana-Cabrera; Mauro Corrado; Sara Cipolat; Veronica Costa; Alberto Casarin; Ligia C. Gomes; et al.Ester Perales-ClementeLeonardo SalviatiPatricio Fernández SilvaJosé Antonio EnríquezLuca Scorrano Mitochondrial Cristae Shape Determines Respiratory Chain Supercomplexes Assembly and Respiratory Efficiency. Cell 2013, 155, 160-171, 10.1016/j.cell.2013.08.032.
- Justine Lebeau; Jaclyn M. Saunders; Vivian W.R. Moraes; Aparajita Madhavan; Nicole Madrazo; Mary C. Anthony; R. Luke Wiseman; The PERK Arm of the Unfolded Protein Response Regulates Mitochondrial Morphology during Acute Endoplasmic Reticulum Stress. Cell Reports 2018, 22, 2827-2836, 10.1016/j.celrep.2018.02.055.
- Laurent Chatel-Chaix; Mirko Cortese; Inés Romero Brey; Silke Bender; Christopher John Neufeldt; Wolfgang Fischl; Pietro Scaturro; Nicole L. Schieber; Yannick Schwab; Bernd Fischer; et al.Alessia RuggieriRalf Bartenschlager Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host & Microbe 2016, 20, 342-356, 10.1016/j.chom.2016.07.008.
- Chong-Shan Shi; Hai-Yan Qi; Cedric Boularan; Ning-Na Huang; Mones Abu-Asab; James H. Shelhamer; John H. Kehrl; SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. The Journal of Immunology 2014, 193, 3080-3089, 10.4049/jimmunol.1303196.
- Krystal A. Fontaine; Roman Camarda; Michael Lagunoff; Vaccinia Virus Requires Glutamine but Not Glucose for Efficient Replication. Journal of Virology 2014, 88, 4366-4374, 10.1128/jvi.03134-13.
- Juan Fontana; Carmen López-Iglesias; Wen-Ping Tzeng; Teryl K. Frey; Jose-Jesus Fernandez; Cristina Risco Ortiz; Three-dimensional structure of Rubella virus factories. Virology 2010, 405, 579-591, 10.1016/j.virol.2010.06.043.
- Juan Fontana; Noelia Lopez-Montero; Richard M. Elliott; José Jesús Fernández; Cristina Risco Ortiz; The unique architecture of Bunyamwera virus factories around the Golgi complex. Cellular Microbiology 2008, 10, 2012-2028, 10.1111/j.1462-5822.2008.01184.x.
- T. Kelly Rainbolt; Jaclyn M. Saunders; R. Luke Wiseman; Stress-responsive regulation of mitochondria through the ER unfolded protein response. Trends in Endocrinology & Metabolism 2014, 25, 528-537, 10.1016/j.tem.2014.06.007.
- David Chan; Fusion and Fission: Interlinked Processes Critical for Mitochondrial Health. Annual Review of Genetics 2012, 46, 265-287, 10.1146/annurev-genet-110410-132529.
- Céline Castanier; Dominique Garcin; Aimé Vazquez; Damien Arnoult; Mitochondrial dynamics regulate the RIG‐I‐like receptor antiviral pathway. EMBO reports 2009, 11, 133-138, 10.1038/embor.2009.258.
- K. Yasukawa; H. Oshiumi; Makoto Takeda; N. Ishihara; Yusuke Yanagi; T. Seya; S.-I. Kawabata; Takumi Koshiba; Mitofusin 2 Inhibits Mitochondrial Antiviral Signaling. Science Signaling 2009, 2, ra47, 10.1126/scisignal.2000287.
- Kazuhide Onoguchi; Koji Onomoto; Shiori Takamatsu; Michihiko Jogi; Azumi Takemura; Shiho Morimoto; Ilkka Julkunen; Hideo Namiki; Mitsutoshi Yoneyama; Takashi Fujita; et al. Virus-Infection or 5′ppp-RNA Activates Antiviral Signal through Redistribution of IPS-1 Mediated by MFN1. PLOS Pathogens 2010, 6, e1001012, 10.1371/journal.ppat.1001012.
- Kawai, T.; Akira, S; Innate immune recognition of viral infection. Nature immunology 2006, 7, 131-137.
- Denis Martinvalet; The role of the mitochondria and the endoplasmic reticulum contact sites in the development of the immune responses. Cell Death & Disease 2018, 9, 336, 10.1038/s41419-017-0237-7.
- Takuma Yoshizumi; Takeshi Ichinohe; Osamu Sasaki; Hidenori Otera; Shun-Ichiro Kawabata; Katsuyoshi Mihara; Takumi Koshiba; Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nature Communications 2014, 5, 4713, 10.1038/ncomms5713.
- Mao Xia; Patrick Gonzalez; Chunyan Li; Gang Meng; Aiqin Jiang; Hongwei Wang; Qian Gao; Klaus-Michael Debatin; Christian Beltinger; Jiwu Wei; et al. Mitophagy Enhances Oncolytic Measles Virus Replication by Mitigating DDX58/RIG-I-Like Receptor Signaling. Journal of Virology 2014, 88, 5152-5164, 10.1128/jvi.03851-13.
- Seong-Jun Kim; Gulam Syed; Mohsin Khan; Wei-Wei Chiu; Muhammad A. Sohail; Robert Gish; Aleem Siddiqui; Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proceedings of the National Academy of Sciences 2014, 111, 6413-6418, 10.1073/pnas.1321114111.
- Seong-Jun Kim; Gulam Syed; Aleem Siddiqui; Hepatitis C Virus Induces the Mitochondrial Translocation of Parkin and Subsequent Mitophagy. PLOS Pathogens 2013, 9, e1003285, 10.1371/journal.ppat.1003285.
- Seong-Jun Kim; Mohsin Khan; Jun Quan; Andreas Till; Suresh Subramani; Aleem Siddiqui; Hepatitis B Virus Disrupts Mitochondrial Dynamics: Induces Fission and Mitophagy to Attenuate Apoptosis. PLOS Pathogens 2013, 9, e1003722, 10.1371/journal.ppat.1003722.
- Marlène Dreux; Pablo Gastaminza; Stefan F. Wieland; Francis V. Chisari; The autophagy machinery is required to initiate hepatitis C virus replication. Proceedings of the National Academy of Sciences 2009, 106, 14046-14051, 10.1073/pnas.0907344106.
- Inés Romero Brey; Ralf Bartenschlager; Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160, 10.3390/v8060160.
- Robert E. Powers; Songyu Wang; Tina Y. Liu; Tom A Rapoport; Reconstitution of the tubular endoplasmic reticulum network with purified components. Nature 2017, 543, 257-260, 10.1038/nature21387.
- Jeanmarie Verchot; How does the stressed out ER find relief during virus infection?. Current Opinion in Virology 2016, 17, 74-79, 10.1016/j.coviro.2016.01.018.
- Miao Wang; Randal J. Kaufman; Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326-335, 10.1038/nature17041.
- E. Sergio Trombetta; Armando J. Parodi; Quality Control and Protein Folding in the Secretory Pathway. Annual Review of Cell and Developmental Biology 2003, 19, 649-676, 10.1146/annurev.cellbio.19.110701.153949.
- Feng-Jin Guo; Zhangyuan Xiong; Xiaojie Lu; Mengliang Ye; Xiaofeng Han; Rong Jiang; ATF6 upregulates XBP1S and inhibits ER stress-mediated apoptosis in osteoarthritis cartilage. Cellular Signalling 2014, 26, 332-342, 10.1016/j.cellsig.2013.11.018.
- Peter Walter; David Ron; The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081-1086, 10.1126/science.1209038.
- Eriko Fujita; Yoriko Kouroku; Atsushi Isoai; Hiromichi Kumagai; Akifumi Misutani; Chie Matsuda; Yukiko K. Hayashi; T Momoi; Two endoplasmic reticulum-associated degradation (ERAD) systems for the novel variant of the mutant dysferlin: ubiquitin/proteasome ERAD(I) and autophagy/lysosome ERAD(II). Human Molecular Genetics 2007, 16, 618-629, 10.1093/hmg/ddm002.
- Stephanie L. Moon; Michael D. Barnhart; Jeffrey Wilusz; Inhibition and avoidance of mRNA degradation by RNA viruses. Current Opinion in Microbiology 2012, 15, 500-505, 10.1016/j.mib.2012.04.009.
- Nai-Yun Hsu; Olga Ilnytska; George A. Belov; Marianita Santiana; Ying-Han Chen; Peter M. Takvorian; Cyrilla Pau; Hilde Van Der Schaar; Neerja Kaushik-Basu; Tamas Balla; et al.Craig E. CameronEllie EhrenfeldFrank J.M. Van KuppeveldNihal Altan-Bonnet Viral Reorganization of the Secretory Pathway Generates Distinct Organelles for RNA Replication. Cell 2010, 141, 799-811, 10.1016/j.cell.2010.03.050.
- Tenorio, R.; Fernandez de Castro, I.; Knowlton, J. J.; Zamora, P. F.; Lee, C. H.; Mainou, B. A.; Dermody, T. S.; Risco, C; Reovirus sigmaNS and muNS Proteins Remodel the Endoplasmic Reticulum to Build Replication Neo-Organelles. mBio 2018, 9, -.
- Sven Miller; Stefan Kastner; Jacomine Krijnse-Locker; Sandra Bühler; Ralf Bartenschlager; The Non-structural Protein 4A of Dengue Virus Is an Integral Membrane Protein Inducing Membrane Alterations in a 2K-regulated Manner. Journal of Biological Chemistry 2007, 282, 8873-8882, 10.1074/jbc.m609919200.
- Cristina Risco Ortiz; Juan R. Rodríguez; Carmen López-Iglesias; José L. Carrascosa; Mariano Esteban; Dolores Rodríguez; Endoplasmic Reticulum-Golgi Intermediate Compartment Membranes and Vimentin Filaments Participate in Vaccinia Virus Assembly. Journal of Virology 2002, 76, 1839-1855, 10.1128/jvi.76.4.1839-1855.2002.
- Matloob Husain; Bernard Moss; Evidence against an Essential Role of COPII-Mediated Cargo Transport to the Endoplasmic Reticulum-Golgi Intermediate Compartment in the Formation of the Primary Membrane of Vaccinia Virus. Journal of Virology 2003, 77, 11754-11766, 10.1128/jvi.77.21.11754-11766.2003.
- Kèvin Knoops; Montserrat Bárcena; Ronald W.A.L. Limpens; Abraham J. Koster; A. Mieke Mommaas; Eric J. Snijder; Ultrastructural Characterization of Arterivirus Replication Structures: Reshaping the Endoplasmic Reticulum To Accommodate Viral RNA Synthesis. Journal of Virology 2011, 86, 2474-2487, 10.1128/jvi.06677-11.
- David A. Suhy; Thomas H. Giddings; Karla Kirkegaard; Remodeling the Endoplasmic Reticulum by Poliovirus Infection and by Individual Viral Proteins: an Autophagy-Like Origin for Virus-Induced Vesicles. Journal of Virology 2000, 74, 8953-8965, 10.1128/jvi.74.19.8953-8965.2000.
- Javier M. Rodríguez; Ramon Garcia-Escudero; María L. Salas; Germán Andrés3; African Swine Fever Virus Structural Protein p54 Is Essential for the Recruitment of Envelope Precursors to Assembly Sites. Journal of Virology 2004, 78, 4299-4313, 10.1128/jvi.78.8.4299-4313.2004.
- Peng Gao; Yue Chai; Jiangwei Song; Teng Liu; Peng Chen; Lei Zhou; Xinna Ge; Xin Guo; Jun Han; Hanchun Yang; et al. Reprogramming the unfolded protein response for replication by porcine reproductive and respiratory syndrome virus. PLOS Pathogens 2019, 15, e1008169, 10.1371/journal.ppat.1008169.
- Shanhui Ren; Zaib Ur Rehman; Mengyu Shi; Bin Yang; Panrao Liu; Yuncong Yin; Yurong Qu; Chunchun Meng; Zengqi Yang; Xiaolong Gao; et al.Yingjie SunChan Ding Hemagglutinin-neuraminidase and fusion proteins of virulent Newcastle disease virus cooperatively disturb fusion–fission homeostasis to enhance mitochondrial function by activating the unfolded protein response of endoplasmic reticulum and mitochondrial stress. Veterinary Research 2019, 50, 37, 10.1186/s13567-019-0654-y.
- Paul Targett-Adams; Steeve Boulant; Mark W. Douglas; John McLauchlan Mclauchlan; Lipid Metabolism and HCV Infection. Viruses 2010, 2, 1195-1217, 10.3390/v2051195.
- Christian Cobbold; Sharon M Brookes; Thomas Wileman; Biochemical Requirements of Virus Wrapping by the Endoplasmic Reticulum: Involvement of ATP and Endoplasmic Reticulum Calcium Store during Envelopment of African Swine Fever Virus. Journal of Virology 2000, 74, 2151-2160, 10.1128/jvi.74.5.2151-2160.2000.
- Liu, X.; Palaniyandi, S.; Zhu, I.; Tang, J.; Li, W.; Wu, X.; Ochsner, S. P.; Pauza, C. D.; Cohen, J. I.; Zhu, X; et al. Human cytomegalovirus evades antibody-mediated immunity through endoplasmic reticulum-associated degradation of the FcRn receptor. Nature communications 2009, 10, 3020.
- Kwang Il Jung; Dong-Hyun Ko; Nary Shin; Chul Woong Pyo; Sang-Yun Choi; Endoplasmic reticulum-associated degradation potentiates the infectivity of influenza A virus by regulating the host redox state. Free Radical Biology and Medicine 2019, 135, 293-305, 10.1016/j.freeradbiomed.2019.03.021.
- Qi Chen; Lijun Sun; Zhijian J. Chen; Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nature Immunology 2016, 17, 1142-1149, 10.1038/ni.3558.
- Mona Motwani; Scott Pesiridis; Katherine A. Fitzgerald; DNA sensing by the cGAS-STING pathway in health and disease. Nature Reviews Microbiology 2019, 20, 657-674, 10.1038/s41576-019-0151-1.
- Ying-Ray Lee; Szu-Han Kuo; Ching-Yen Lin; Po-Jung Fu; Yee-Shin Lin; Trai-Ming Yeh; Hsiao-Sheng Liu; Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Scientific Reports 2018, 8, 489, 10.1038/s41598-017-18909-3.
- David Ron; Peter Walter; Signal integration in the endoplasmic reticulum unfolded protein response. Nature Reviews Molecular Cell Biology 2007, 8, 519-529, 10.1038/nrm2199.
- Ying-Ray Lee; Szu-Han Kuo; Ching-Yen Lin; Po-Jung Fu; Yee-Shin Lin; Trai-Ming Yeh; Hsiao-Sheng Liu; Dengue virus-induced ER stress is required for autophagy activation, viral replication, and pathogenesis both in vitro and in vivo. Scientific Reports 2018, 8, 489, 10.1038/s41598-017-18909-3.
- Qianruo Wang; Xiu Xin; Ting Wang; Jiawu Wan; Yangtao Ou; Zibing Yang; Qijia Yu; Liting Zhu; Yunli Guo; Yinsheng Wu; et al.Zhen DingYanni ZhangZishu PanYuxin TangShanshan LiLingbao Kong Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. Journal of Virology 2019, 93, -, 10.1128/jvi.00887-19.
- Mingjie Huang; Ahui Xu; Xiaoyu Wu; Yanni Zhang; Yunli Guo; Fenglin Guo; Zishu Pan; Lingbao Kong; Japanese encephalitis virus induces apoptosis by the IRE1/JNK pathway of ER stress response in BHK-21 cells. Archives of Virology 2015, 161, 699-703, 10.1007/s00705-015-2715-5.
- Manish Sharma; Sankar Bhattacharyya; Kiran Bala Sharma; Shailendra Chauhan; Suramya Asthana; Malik Zainul Abdin; Sudhanshu Vrati; Manjula Kalia; Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. Journal of General Virology 2017, 98, 1027-1039, 10.1099/jgv.0.000792.
- Chao Yu; Katharina Achazi; Matthias Niedrig; Tick-borne encephalitis virus triggers inositol-requiring enzyme 1 (IRE1) and transcription factor 6 (ATF6) pathways of unfolded protein response. Virus Research 2013, 178, 471-477, 10.1016/j.virusres.2013.10.012.
- Keith D. Tardif; Kazutoshi Mori; Randal J. Kaufman; Aleem Siddiqui; Hepatitis C Virus Suppresses the IRE1-XBP1 Pathway of the Unfolded Protein Response. Journal of Biological Chemistry 2004, 279, 17158-17164, 10.1074/jbc.m312144200.
- Ihab Hassan; Michael S. Zhang; Linda S. Powers; Jian Q. Shao; Jonas Baltrusaitis; D. Thomas Rutkowski; Kevin L. Legge; Martha M. Monick; Influenza A Viral Replication Is Blocked by Inhibition of the Inositol-requiring Enzyme 1 (IRE1) Stress Pathway. Journal of Biological Chemistry 2011, 287, 4679-4689, 10.1074/jbc.M111.284695.
- Li, Y.; Jiang, W.; Niu, Q.; Sun, Y.; Meng, C.; Tan, L.; Song, C.; Qiu, X.; Liao, Y.; Ding, C; et al. eIF2alpha-CHOP-BCl-2/JNK and IRE1alpha-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell death & disease 2019, 10, 891.
- Sebastian Stahl; Julia M. Burkhart; Florian Hinte; Boaz Tirosh; Hermine Mohr; René P. Zahedi; Albert Sickmann; Zsolt Ruzsics; Matthias Budt; Wolfram Brune; et al. Cytomegalovirus Downregulates IRE1 to Repress the Unfolded Protein Response. PLOS Pathogens 2013, 9, e1003544, 10.1371/journal.ppat.1003544.
- Zhikang Qian; Baoqin Xuan; Travis J. Chapa; Nathaniel Gualberto; Dong Yu; Murine Cytomegalovirus Targets Transcription Factor ATF4 To Exploit the Unfolded-Protein Response. Journal of Virology 2012, 86, 6712-6723, 10.1128/jvi.00200-12.
- Heather F. Burnett; Timothy E Audas; Genqing Liang; Ray Lu; Herpes simplex virus-1 disarms the unfolded protein response in the early stages of infection. Cell Stress and Chaperones 2012, 17, 473-483, 10.1007/s12192-012-0324-8.
- Pengchao Zhang; Chenhe Su; Zhangtao Jiang; Chunfu Zheng; Herpes Simplex Virus 1 UL41 Protein Suppresses the IRE1/XBP1 Signal Pathway of the Unfolded Protein Response via Its RNase Activity. Journal of Virology 2016, 91, e02056-16, 10.1128/jvi.02056-16.
- Cheng, G.; Feng, Z.; He, B; Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. Journal of virology 2005, 79, 1379-1388.
- Matthew Mulvey; Carolina Arias; Ian Mohr; Maintenance of Endoplasmic Reticulum (ER) Homeostasis in Herpes Simplex Virus Type 1-Infected Cells through the Association of a Viral Glycoprotein with PERK, a Cellular ER Stress Sensor. Journal of Virology 2007, 81, 3377-3390, 10.1128/jvi.02191-06.
- Baozong Li; Bo Gao; Linbai Ye; Xue Han; Wei Wang; Lingbao Kong; Xiaonan Fang; Yingchun Zeng; Hong Zheng; Shanshan Li; et al.Zhenghui WuLi Ye Hepatitis B virus X protein (HBx) activates ATF6 and IRE1-XBP1 pathways of unfolded protein response. Virus Research 2007, 124, 44-49, 10.1016/j.virusres.2006.09.011.
- Ronald Wanders; Hans R. Waterham; Biochemistry of Mammalian Peroxisomes Revisited. Annual Review of Biochemistry 2006, 75, 295-332, 10.1146/annurev.biochem.74.082803.133329.
- Till, A.; Lakhani, R.; Burnett, S. F.; Subramani, S; Pexophagy: The Selective Autophagy of Peroxisomes. International journal of cell biology 2012, 2012, 512721.
- Honsho, M.; Yamashita, S.-i.; Fujiki, Y; Pexophagy: The Selective Autophagy of PeroxisomesPeroxisome homeostasis: Mechanisms of division and selective degradation of peroxisomes in mammals. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 2016, 1863, 984-991.
- Hannah K. Delille; Michael Schrader; Targeting of hFis1 to Peroxisomes Is Mediated by Pex19p. Journal of Biological Chemistry 2008, 283, 31107-31115, 10.1074/jbc.M803332200.
- Michael Schrader; Shared components of mitochondrial and peroxisomal division. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2006, 1763, 531-541, 10.1016/j.bbamcr.2006.01.004.
- Margaret Neuspiel; Astrid C. Schauss; Emelie Braschi; Rodolfo Zunino; Peter Rippstein; Richard A. Rachubinski; Miguel A Andrade-Navarro; Heidi M McBride; Cargo-Selected Transport from the Mitochondria to Peroxisomes Is Mediated by Vesicular Carriers. Current Biology 2008, 18, 102-108, 10.1016/j.cub.2007.12.038.
- Emelie Braschi; Vanessa Goyon; Rodolfo Zunino; Abhishek Mohanty; Liqun Xu; Heidi M McBride; Vps35 Mediates Vesicle Transport between the Mitochondria and Peroxisomes. Current Biology 2010, 20, 1310-1315, 10.1016/j.cub.2010.05.066.
- K. V. K. Mohan; I. Som; Chintamani D. Atreya; Identification of a Type 1 Peroxisomal Targeting Signal in a Viral Protein and Demonstration of Its Targeting to the Organelle. Journal of Virology 2002, 76, 2543-2547, 10.1128/jvi.76.5.2543-2547.2002.
- Begoña Aguado; Eladio Vañuela; Antonio Alcamí; African swine fever virus fatty acid acylated proteins. Virology 1991, 185, 942-945, 10.1016/0042-6822(91)90578-y.
- Andrew W. McCartney; John S. Greenwood; Marc R. Fabian; K. Andrew White; Robert T. Mullen; Localization of the Tomato Bushy Stunt Virus Replication Protein p33 Reveals a Peroxisome-to-Endoplasmic Reticulum Sorting PathwayW. The Plant Cell 2005, 17, 3513-3531, 10.1105/tpc.105.036350.
- Magdalena Jonczyk; Kunj B. Pathak; Monika Sharma; Peter D. Nagy; Exploiting alternative subcellular location for replication: Tombusvirus replication switches to the endoplasmic reticulum in the absence of peroxisomes. Virology 2007, 362, 320-330, 10.1016/j.virol.2007.01.004.
- D. James Morré; Hilton H. Mollenhauer; Microscopic Morphology and the Origins of the Membrane Maturation Model of Golgi Apparatus Function. Natural and Engineered Resistance to Plant Viruses, Part II 2007, 262, 191-218, 10.1016/s0074-7696(07)62004-x.
- C Barlowe; COPII: A membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 1994, 77, 895-907, 10.1016/0092-8674(94)90138-4.
- Rabouille, C.; Klumperman, J; Opinion: The maturing role of COPI vesicles in intra-Golgi transport. Nature reviews. Molecular cell biology 2005, 6, 812-817.
- George A. Belov; Nihal Altan-Bonnet; Gennadiy Kovtunovych; Catherine L. Jackson; Jennifer Lippincott-Schwartz; Ellie Ehrenfeld; Hijacking Components of the Cellular Secretory Pathway for Replication of Poliovirus RNA. Journal of Virology 2006, 81, 558-567, 10.1128/jvi.01820-06.
- René C. Rust; Lukas Landmann; Rainer Gosert; Bor Luen Tang; Wanjin Hong; Hans-Peter Hauri; Denise Egger; Kurt Bienz; Cellular COPII Proteins Are Involved in Production of the Vesicles That Form the Poliovirus Replication Complex. Journal of Virology 2001, 75, 9808-9818, 10.1128/jvi.75.20.9808-9818.2001.
- J.R. Doedens; K. Kirkegaard; Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. The EMBO Journal 1995, 14, 894-907, 10.1002/j.1460-2075.1995.tb07071.x.
- Dodd, D. A.; Giddings, T. H., Jr.; Kirkegaard, K; Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. Journal of virology 2001, 75, 8158-8165.
- Stephen B. Deitz; Dana A. Dodd; Stewart Cooper; Peter Parham; Karla Kirkegaard; MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proceedings of the National Academy of Sciences 2000, 97, 13790-13795, 10.1073/pnas.250483097.
- R. García-Beato; M.L. Salas; E. Viñuela; J. Salas; Role of the host cell nucleus in the replication of African swine fever virus DNA. Virology 1992, 188, 637-649, 10.1016/0042-6822(92)90518-t.
- Dixon, L. K.; Chapman, D. A.; Netherton, C. L.; Upton, C; African swine fever virus replication and genomics. Virus research 2013, 173, 3-14.
- George C. Katsafanas; Bernard Moss; Colocalization of Transcription and Translation within Cytoplasmic Poxvirus Factories Coordinates Viral Expression and Subjugates Host Functions. Cell Host & Microbe 2007, 2, 221-228, 10.1016/j.chom.2007.08.005.
- Ketil W. Pedersen; Yvonne Van Der Meer; Norbert Roos; Eric J. Snijder; Open Reading Frame 1a-Encoded Subunits of the Arterivirus Replicase Induce Endoplasmic Reticulum-Derived Double-Membrane Vesicles Which Carry the Viral Replication Complex. Journal of Virology 1999, 73, 2016-2026.
- Megan M. Angelini; Marzieh Akhlaghpour; Benjamin W. Neuman; Michael J. Buchmeier; Severe Acute Respiratory Syndrome Coronavirus Nonstructural Proteins 3, 4, and 6 Induce Double-Membrane Vesicles. mBio 2013, 4, e00524-13, 10.1128/mbio.00524-13.
- Anne G. Bost; Erik Prentice; Mark R. Denison; Mouse Hepatitis Virus Replicase Protein Complexes Are Translocated to Sites of M Protein Accumulation in the ERGIC at Late Times of Infection. Virology 2001, 285, 21-29, 10.1006/viro.2001.0932.
- Silke Stertz; Mike Reichelt; Martin Spiegel; Thomas Kuri; Luis Martínez-Sobrido; Adolfo García-Sastre; Friedemann Weber; Georg Kochs; The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304-315, 10.1016/j.virol.2006.11.027.
- Linya Wang; Yongjun Tian; Jing-Hsiung James Ou; HCV Induces the Expression of Rubicon and UVRAG to Temporally Regulate the Maturation of Autophagosomes and Viral Replication. PLOS Pathogens 2015, 11, e1004764, 10.1371/journal.ppat.1004764.
- Nna Sir; Cheng-Fu Kuo; Yongjun Tian; Helene Minyi Liu; Eric J. Huang; Jae U. Jung; Keigo Machida; Jing-Hsiung James Ou; Replication of Hepatitis C Virus RNA on Autophagosomal Membranes. Journal of Biological Chemistry 2012, 287, 18036-18043, 10.1074/jbc.M111.320085.
- Bjorn-Patrick Mohl; Christopher Bartlett; Jamel Mankouri; Mark Harris; Early events in the generation of autophagosomes are required for the formation of membrane structures involved in hepatitis C virus genome replication. Journal of General Virology 2016, 97, 680-693, 10.1099/jgv.0.000387.
- Dianna Magliano; John A. Marshall; D.Scott Bowden; Nicholas Vardaxis; Jayesh Meanger; Jia-Yee Lee; Rubella Virus Replication Complexes Are Virus-Modified Lysosomes. Virology 1998, 240, 57-63, 10.1006/viro.1997.8906.
- David J. Miller; Michael D. Schwartz; Paul Ahlquist; Flock House Virus RNA Replicates on Outer Mitochondrial Membranes in Drosophila Cells. Journal of Virology 2001, 75, 11664-11676, 10.1128/jvi.75.23.11664-11676.2001.
- Kenneth J Ertel; Desirée Benefield; Daniel Castaño-Diez; Janice G Pennington; Mark Horswill; Johan A Den Boon; Marisa S Otegui; Paul Ahlquist; Cryo-electron tomography reveals novel features of a viral RNA replication compartment. eLife 2017, 6, -, 10.7554/eLife.25940.
- Pekka Kujala; Anne Ikäheimonen; Neda Ehsani; Helena Vihinen; Petri Auvinen; L. Kääriäinen; Biogenesis of the Semliki Forest Virus RNA Replication Complex. Journal of Virology 2001, 75, 3873-3884, 10.1128/jvi.75.8.3873-3884.2001.
- S Froshauer; J Kartenbeck; A Helenius; Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. Journal of Cell Biology 1988, 107, 2075-2086.
- Pirjo Spuul; Giuseppe Balistreri; Leevi Kääriäinen; Tero Ahola; Phosphatidylinositol 3-Kinase-, Actin-, and Microtubule-Dependent Transport of Semliki Forest Virus Replication Complexes from the Plasma Membrane to Modified Lysosomes. Journal of Virology 2010, 84, 7543-7557, 10.1128/jvi.00477-10.
- Jim Baggen; Hendrik Jan Thibaut; Jeroen R. P. M. Strating; Frank J.M. Van Kuppeveld; The life cycle of non-polio enteroviruses and how to target it. Nature Reviews Microbiology 2018, 16, 368-381, 10.1038/s41579-018-0005-4.
- Jason S. Long; Bhakti Mistry; S. M. Haslam; Wendy S. Barclay; Host and viral determinants of influenza A virus species specificity. Nature Reviews Microbiology 2019, 17, 67-81, 10.1038/s41579-018-0115-z.
- Xiǎohóng Shí; Alain Kohl; Vincent H. J. Léonard; Ping Li; Angela McLees; Richard M. Elliott; Requirement of the N-Terminal Region of Orthobunyavirus Nonstructural Protein NSm for Virus Assembly and Morphogenesis. Journal of Virology 2006, 80, 8089-8099, 10.1128/jvi.00579-06.
- Candace Gomez; Thomas J. Hope; The ins and outs of HIV replication. Cellular Microbiology 2005, 7, 621-626, 10.1111/j.1462-5822.2005.00516.x.
- Sonja Aits; Marja Jäättelä; Lysosomal cell death at a glance. Journal of Cell Science 2013, 126, 1905-1912, 10.1242/jcs.091181.
- Rushika M. Perera; Roberto Zoncu; The Lysosome as a Regulatory Hub. Annual Review of Cell and Developmental Biology 2016, 32, 223-253, 10.1146/annurev-cellbio-111315-125125.
- Paul Saftig; Judith Klumperman; Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nature Reviews Molecular Cell Biology 2009, 10, 623-635, 10.1038/nrm2745.
- Issam Tout; Melissa Gomes; Michelle Ainouze; Marie Marotel; Timothee Pecoul; David Durantel; Salvatore Vaccarella; Bertrand Dubois; Veronique Loustaud-Ratti; Thierry Walzer; et al.Sophie AlainIsabelle CheminUzma Hasan Hepatitis B Virus Blocks the CRE/CREB Complex and Prevents TLR9 Transcription and Function in Human B Cells. The Journal of Immunology 2018, 201, 2331-2344, 10.4049/jimmunol.1701726.
- Jenn-Haung Lai; Mei‐Yi Wang; Chuan‐Yueh Huang; Chien‐Hsiang Wu; Li‐Feng Hung; Chia‐Ying Yang; Po-Yuan Ke; Shue‐Fen Luo; Shih‐Jen Liu; Ling-Jun Ho; et al. Infection with the dengue RNA virus activates TLR9 signaling in human dendritic cells. EMBO reports 2018, 19, e46182, 10.15252/embr.201846182.
- Juan S. Bonifacino; Linton Traub; Signals for Sorting of Transmembrane Proteins to Endosomes and Lysosomes. Annual Review of Biochemistry 2003, 72, 395-447, 10.1146/annurev.biochem.72.121801.161800.