The immune microenvironment of high-grade gliomas (HGG) is a complex and heterogeneous system, consisting of diverse cell types such as microglia, bone marrow-derived macrophages (BMDMs), myeloid-derived suppressor cells (MDSCs), dendritic cells, natural killer (NK) cells, and T-cells. Of these, MDSCs are one of the major tumor-infiltrating immune cells and are correlated not only with overall worse prognosis but also poor clinical outcomes. Upon entry from the bone marrow into the peripheral blood, spleen, as well as in tumor microenvironment (TME) in HGG patients, MDSCs deploy an array of mechanisms to perform their immune and non-immune suppressive functions.
- MDSCs
- glioma
- glioblastoma
- high-grade glioma
- brain tumors
- metabolic reprogramming
- immune suppression
- therapeutic resistance
- therapeutic targeting
- immunotherapy
- tumor microenvironment
1. Introduction
Extensive analysis of the immune microenvironment in high-grade glioma (HGG) using single-cell RNA-seq, mass cytometry (CyTOF), immunohistochemistry, flow cytometry, and other “omics” technologies indicate the presence of higher numbers of immune-suppressive macrophages, microglia dendritic cells, regulatory T-cells, and myeloid-derived suppressor cells (MDSCs) [5]. Together, these cells interact with the neoplastic cells to promote tumor growth, progression, metastasis, angiogenesis and contribute to the extreme immunosuppression observed in HGG.
Extensive analysis of the immune microenvironment in high-grade glioma (HGG) using single-cell RNA-seq, mass cytometry (CyTOF), immunohistochemistry, flow cytometry, and other “omics” technologies indicate the presence of higher numbers of immune-suppressive macrophages, microglia dendritic cells, regulatory T-cells, and myeloid-derived suppressor cells (MDSCs) [1]. Together, these cells interact with the neoplastic cells to promote tumor growth, progression, metastasis, angiogenesis and contribute to the extreme immunosuppression observed in HGG.
In healthy humans and mice, MDSCs are present at very low frequencies and constitute only ~0.5–2% of peripheral blood mononuclear cells [6]. Nevertheless, 30–50% of the tumor mass in HGGs are found to be MDSCs [7,8]. Originally, derived from the bone marrow, MDSCs are a very heterogeneous population of immature myeloid cells (IMCs) present at various stages of myelopoiesis. Under normal conditions, IMCs can be differentiated into macrophages, granulocytes, and dendritic cells. However, in pathological conditions such as HGG, the differentiation of IMCs is subverted, resulting in the generation, recruitment, expansion, and activation of MDSCs [9] not only in the tumor bed but also in the peripheral blood [10,11].
In healthy humans and mice, MDSCs are present at very low frequencies and constitute only ~0.5–2% of peripheral blood mononuclear cells [2]. Nevertheless, 30–50% of the tumor mass in HGGs are found to be MDSCs [3][4]. Originally, derived from the bone marrow, MDSCs are a very heterogeneous population of immature myeloid cells (IMCs) present at various stages of myelopoiesis. Under normal conditions, IMCs can be differentiated into macrophages, granulocytes, and dendritic cells. However, in pathological conditions such as HGG, the differentiation of IMCs is subverted, resulting in the generation, recruitment, expansion, and activation of MDSCs [5] not only in the tumor bed but also in the peripheral blood [6][7].
2. History of MDSCs
In HGGs, MDSCs are derived from the immature myeloid progenitors present in the bone marrow (
Figure 1). More recently, single-cell RNA-sequencing (scRNA-seq) analysis on mouse breast tumors suggests that abnormal differentiation of monocyte and neutrophil-like cells in the spleen can lead to the generation of MDSCs [17]. Reprogramming or activation of monocytes and granulocytes by exposure to Toll-like receptors, IL-10, WNT5A, LPS, and INFγ can also give rise to MDSCs [18]. Lastly, MDSCs can be generated and activated ex vivo by the addition of GM-CSF, G-CSF, IL-6, and IL-10 to bone marrow precursors obtained from healthy individuals and detailed functional and phenotypic characterization revealed these bone marrow-derived MDSCs (BM-MDSCs) to be similar to the MDSCs present in different cancer patients [19,20].
). More recently, single-cell RNA-sequencing (scRNA-seq) analysis on mouse breast tumors suggests that abnormal differentiation of monocyte and neutrophil-like cells in the spleen can lead to the generation of MDSCs [8]. Reprogramming or activation of monocytes and granulocytes by exposure to Toll-like receptors, IL-10, WNT5A, LPS, and INFγ can also give rise to MDSCs [9]. Lastly, MDSCs can be generated and activated ex vivo by the addition of GM-CSF, G-CSF, IL-6, and IL-10 to bone marrow precursors obtained from healthy individuals and detailed functional and phenotypic characterization revealed these bone marrow-derived MDSCs (BM-MDSCs) to be similar to the MDSCs present in different cancer patients [10][11].
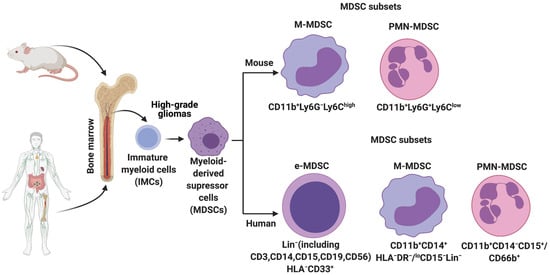
Figure 1.
Origin and subsets of myeloid-derived suppressor cells (MDSCs) in mice and humans. Under normal conditions, immature myeloid cells (IMCs) derived from the bone marrow differentiate into Monocytes, Neutrophils, Macrophages, and Dendritic cells. However, pathological conditions such as high-grade gliomas prevent IMC differentiation, resulting in the generation and accumulation of different MDSC subsets. Phenotypic markers that are commonly used to identify these subsets in mice and humans are described.
3. MDSCs in High-Grade Gliomas
In rat glioma models, an increase in the percentage of MDSCs in the peripheral blood has been reported [33]. Furthermore, it can also serve as a biomarker for tumor recurrence [33]. An increase in MDSCs is also reported in syngeneic and xenograft mouse models of glioma [34]. Corroborating these findings, in humans, elevated levels of MDSCs in the peripheral blood of glioblastoma (GBM) patients have been observed [35], however, this increase is yet to be correlated with increased MDSCs at the glioma site. One possible explanation for this is attributed to the existence of different subsets of MDSCs.
In rat glioma models, an increase in the percentage of MDSCs in the peripheral blood has been reported [12]. Furthermore, it can also serve as a biomarker for tumor recurrence [12]. An increase in MDSCs is also reported in syngeneic and xenograft mouse models of glioma [13]. Corroborating these findings, in humans, elevated levels of MDSCs in the peripheral blood of glioblastoma (GBM) patients have been observed [14], however, this increase is yet to be correlated with increased MDSCs at the glioma site. One possible explanation for this is attributed to the existence of different subsets of MDSCs.
The precise mechanisms by which MDSCs traffic from bone marrow to the gliomas are not known. To recruit MDSCs from the bone marrow, glioma cells overexpress CD200 [39], indoleamine 2,3-dioxygenase (IDO1) [40] and secrete CCL20 [41], macrophage migration inhibitory factor (MIF) [37], in addition to other growth factors. Along with glioma cells, immune cells such as glioma-associated microglia and macrophages (GAMs) secrete CCL2 to recruit regulatory T-cells (Tregs) and MDSCs (
The precise mechanisms by which MDSCs traffic from bone marrow to the gliomas are not known. To recruit MDSCs from the bone marrow, glioma cells overexpress CD200 [15], indoleamine 2,3-dioxygenase (IDO1) [16] and secrete CCL20 [17], macrophage migration inhibitory factor (MIF) [18], in addition to other growth factors. Along with glioma cells, immune cells such as glioma-associated microglia and macrophages (GAMs) secrete CCL2 to recruit regulatory T-cells (Tregs) and MDSCs (
Figure 2). Furthermore, the hypoxic tumor environment upregulates the expression of vascular endothelial growth factor (VEGF) and hypoxia-inducible factor-1 alpha (HIF1α) in glioma cells, which then induces ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2) and aids in the accumulation of MDSCs by converting extracellular ATP to 5′-AMP [42]. In addition, hypoxia recruits CX3CR1 expressing MDSCs by increasing the expression of CCL26 on tumor cells [43]. Finally, in vitro exposure of MDSCs from the spleen to hypoxia results in their differentiation into immunosuppressive macrophages [44].
). Furthermore, the hypoxic tumor environment upregulates the expression of vascular endothelial growth factor (VEGF) and hypoxia-inducible factor-1 alpha (HIF1α) in glioma cells, which then induces ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2) and aids in the accumulation of MDSCs by converting extracellular ATP to 5′-AMP [19]. In addition, hypoxia recruits CX3CR1 expressing MDSCs by increasing the expression of CCL26 on tumor cells [20]. Finally, in vitro exposure of MDSCs from the spleen to hypoxia results in their differentiation into immunosuppressive macrophages [21].
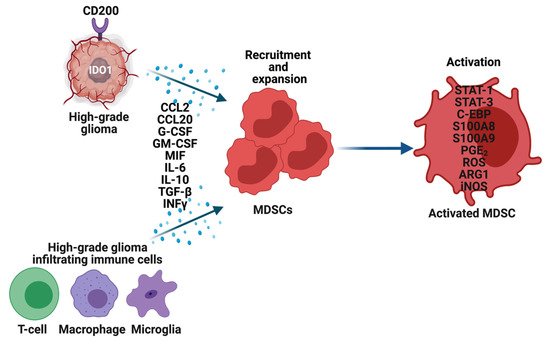
Recruitment, expansion, and activation of MDSCs in high-grade gliomas (HGG). Glioma cells, T-cells, Macrophages, and Microglia in the tumor microenvironment overexpress multiple genes and secrete an array of cytokines, chemokines, and other factors to recruit and expand MDSCs. These factors also activate MDSCs through various mechanisms, which then perform their immune-suppressive functions in HGG.
Once MDSCs are recruited to the HGG site their expansion and activation are tightly controlled by cytokines (IL-6, IL-10, TGF-β, M-CSF, GM-CSF, INFγ), chemokines (CCR2, and other factors (VEGF) secreted by tumor cells, T cells, microglia, and macrophages (
In summary, glioma and immune cells develop and maintain immunosuppressive TME by recruiting, expanding, and activating MDSCs from the bone marrow and spleen into the peripheral blood, as well as at the tumor site.
4. Metabolic Reprogramming of MDSCs
Increased glycolysis, ROS, fatty acid oxidation (FAO), glutamine metabolism, oxidative phosphorylation (OXPHOS), lipid uptake, and extracellular adenosine are observed in MDSCs infiltrating into the tumors, including HGG (
Figure 3) [54]. MDSCs exhibit these metabolic changes to support their development, survival, differentiation, activation, and immunosuppressive activity. Thus, inhibitors targeting the above-mentioned metabolic pathways are being actively explored to block the MDSC activity and improve anti-cancer immunotherapies.
) [23]. MDSCs exhibit these metabolic changes to support their development, survival, differentiation, activation, and immunosuppressive activity. Thus, inhibitors targeting the above-mentioned metabolic pathways are being actively explored to block the MDSC activity and improve anti-cancer immunotherapies.
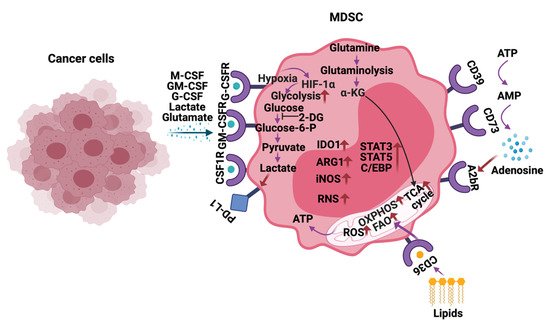
Figure 3.
Metabolic reprogramming of MDSCs in the tumor microenvironment. In response to the cytokines and metabolites secreted by the cancer cells, MDSCs alter their metabolism by increasing glycolysis, glutamine/glutaminolysis, fatty acid oxidation (FAO), oxidative phosphorylation (OXPHOS), tricarboxylic acid (TCA) cycle, reactive oxygen species (ROS), and expression of ectonucleotidases CD39/CD73. Additionally, metabolites such as lactate, lipids glutamate, and adenosine in the tumor microenvironment (TME) also play a vital role in regulating the immune-suppressive functions of MDSCs in HGG. The arrows in red indicate activation/upregulation.
5. Immunosuppression Mediated by MDSCs
In steady-state or homeostatic conditions MDSCs lack any immunosuppressive activity. This is due to the shared characteristics of MDSCs with neutrophils and monocytes. However, in pathological diseases, inflammation, obesity, sepsis, and cancer, immunosuppression is one of the major functions orchestrated by MDSCs. To this end, MDSCs employ a variety of mechanisms to inhibit the function of T-cells, NK, dendritic cells (DC), and macrophages and promote the recruitment of Tregs and immune-suppressive B-cells to support the development and progression of HGG (
). Furthermore, MDSCs generate and maintain an immunosuppressive TME and limit the efficacy of cancer immunotherapies.
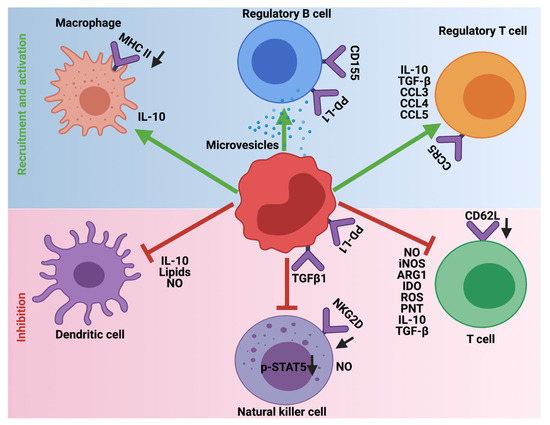
Figure 4.
Immunosuppressive role of MDSCs in HGG. MDSCs employ different mechanisms to inhibit the cytotoxic and antigen presentation functions of T-cells, Natural killer cells, and Dendritic cells. Additionally, MDSCs promote the recruitment and activation of immune-suppressive Tregs, Bregs, and M2 macrophages. The downward arrows in black indicate downregulation.
MDSCs are one of the key players in avoiding immune surveillance in HGG. To achieve this, MDSCs deploy different strategies to prevent the cytotoxic functions and maturation of T-cells, NK cells, and DCs. Additionally, they provide a favorable TME to support the recruitment and inhibitory functions of M2 macrophages, Tregs, and Bregs. Whether some of these outlined mechanisms contribute to the immunosuppression by MDSCs in HGGs remain to be investigated.
6. MDSCs Induced Therapeutic Resistance
MDSCs levels in the peripheral blood as well as at the tumor site are used as a biomarker to predict if the existing standard-of-care therapies would work in cancer patients. High levels of MDSCs are often correlated with chemo, radiation, and immunotherapy resistance (
).
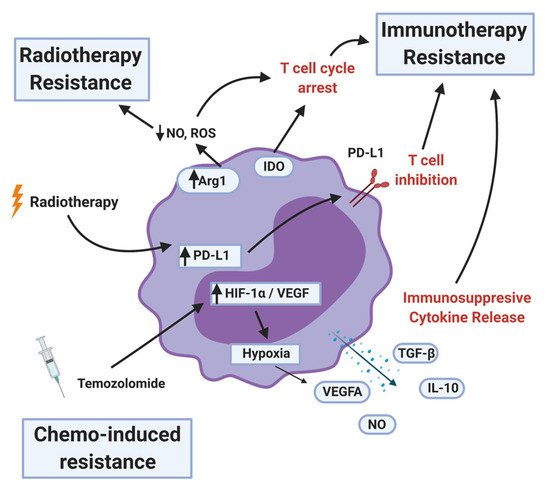
Figure 5.
MDSC mechanisms of therapy resistance. MDSCs contribute to radiotherapy resistance by depleting nitric oxide (NO) and reactive oxygen species (ROS) and upregulating programmed death ligand-1 (PD-L1) after the treatment. MDSCs inhibit immunotherapy by arresting or inhibiting T-cell function, recruiting T-regulatory cells, and secreting immunosuppressive cytokines and other factors. Less is known about chemotherapy resistance, but certain MDSC populations can be a proxy for therapy response. MDSCs that survive the initial temozolomide treatment upregulates hypoxia-inducible factor-1 alpha (HIF-1α) and vascular endothelial growth factor (VEGF), resulting in pro-tumor angiogenesis.
MDSCs represent a large obstacle to the immune checkpoint blockade due to their direct suppression and indirect induction of immunosuppressive phenotypes in other immune cells in the tumor microenvironment. In glioblastoma, MDSCs are influenced by tumor cells, including cancer stem cells, towards their immunosuppressive phenotype both locally and systemically [48,100].
MDSCs represent a large obstacle to the immune checkpoint blockade due to their direct suppression and indirect induction of immunosuppressive phenotypes in other immune cells in the tumor microenvironment. In glioblastoma, MDSCs are influenced by tumor cells, including cancer stem cells, towards their immunosuppressive phenotype both locally and systemically [24][25].
MDSCs secrete immunosuppressive cytokines and other factors such as TGF-β, IL-10, NO, and ROS. They also upregulate proteins that induce T-cell dysfunction: Arg1, IDO1, and PD-L1 (
Figure 5). A further consequence of MDSCs is the induction of immunosuppressive phenotypes in other immune cell types in the TME. MDSCs promote the differentiation of Treg cells [110]. Tregs are potent suppressors of both the adaptive and innate immune system [111]. MDSCs contribute to increased NK cell dysfunction [81] and T-cell exhaustion by inducing T-cell expression of TIGIT, CTLA-4, TIM3, LAG3, and CD160 [112,113]. Whether these effects seen in other tumor types translate to HGG remains to be investigated.
). A further consequence of MDSCs is the induction of immunosuppressive phenotypes in other immune cell types in the TME. MDSCs promote the differentiation of Treg cells [26]. Tregs are potent suppressors of both the adaptive and innate immune system [27]. MDSCs contribute to increased NK cell dysfunction [28] and T-cell exhaustion by inducing T-cell expression of TIGIT, CTLA-4, TIM3, LAG3, and CD160 [29][30]. Whether these effects seen in other tumor types translate to HGG remains to be investigated.
The result of MDSCs in the TME is a self-reinforcing network of immune suppression that supports tumor proliferation. As a result of these known suppressive mechanisms, it is no surprise that the presence of MDSCs can affect the efficacy of IC therapy. In a model of metastatic melanoma, MDSC levels could be used as a predictor of ipilimumab (anti-CTLA4 monoclonal antibody) treatment efficacy [114]. Future treatment models may need to include an anti-MDSC component. Promising research in mouse models has shown rejection of tumor and immunologic memory when combining immune checkpoint inhibitors (nivolumab, avelumab, and ipilimumab) with MDSC targeted therapies as discussed below [115].
The result of MDSCs in the TME is a self-reinforcing network of immune suppression that supports tumor proliferation. As a result of these known suppressive mechanisms, it is no surprise that the presence of MDSCs can affect the efficacy of IC therapy. In a model of metastatic melanoma, MDSC levels could be used as a predictor of ipilimumab (anti-CTLA4 monoclonal antibody) treatment efficacy [31]. Future treatment models may need to include an anti-MDSC component. Promising research in mouse models has shown rejection of tumor and immunologic memory when combining immune checkpoint inhibitors (nivolumab, avelumab, and ipilimumab) with MDSC targeted therapies as discussed below [32].
7. Strategies for Therapeutic Targeting of MDSCs
Several strategies are being developed to therapeutically target MDSCs in solid cancers (
).
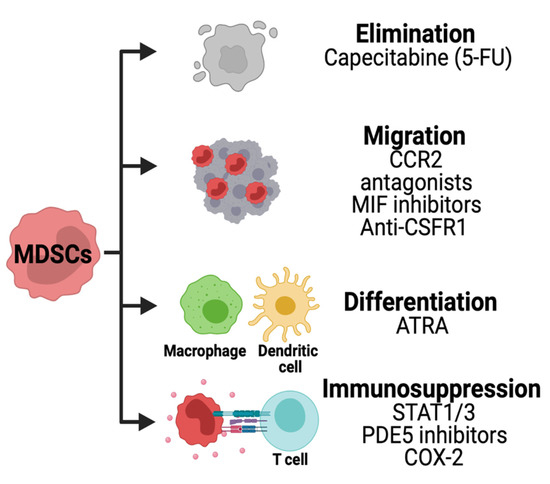
Figure 6.
Strategies for targeting MDSCs in HGG. Chemotherapies are being used to deplete MDSCs in solid tumors. The migration of these cells is inhibited using CCR2, macrophage inhibitory factor (MIF) or CSFR1 inhibitors. Other therapies are used to decrease the immunosuppression pathways and promote MDSCs differentiation.
The immunosuppressive activity of MDSCs can be disrupted by inhibiting stem cell factors primarily produced by tumor cells. The JAK2-STAT3 pathway is known to regulate immune responses via cytokines. JAK2 phosphorylation and activation cause translocation of STAT proteins into the nucleus regulating anti-apoptotic and pro-proliferative genes in MDSCs. A STAT3 inhibitor has been identified from the herb Curcuma longa Linn commonly known as curcumin. Studies have shown the benefits of curcumin in brain tumor cells, decreasing their proliferative and antiapoptotic capacity [118,119]. However, the effects of STAT3 inhibition in MDSCs in the setting of brain tumors remain unexplored to this date.
The immunosuppressive activity of MDSCs can be disrupted by inhibiting stem cell factors primarily produced by tumor cells. The JAK2-STAT3 pathway is known to regulate immune responses via cytokines. JAK2 phosphorylation and activation cause translocation of STAT proteins into the nucleus regulating anti-apoptotic and pro-proliferative genes in MDSCs. A STAT3 inhibitor has been identified from the herb Curcuma longa Linn commonly known as curcumin. Studies have shown the benefits of curcumin in brain tumor cells, decreasing their proliferative and antiapoptotic capacity [33][34]. However, the effects of STAT3 inhibition in MDSCs in the setting of brain tumors remain unexplored to this date.
Phosphodiesterase 5 (PDE5) is another promising therapeutic targeting immunosuppressive pathway in MDSCs. PDE5 is a hydrolase that regulates the NO/cyclic guanosine monophosphate (cGMP) pathway [120].
Phosphodiesterase 5 (PDE5) is another promising therapeutic targeting immunosuppressive pathway in MDSCs. PDE5 is a hydrolase that regulates the NO/cyclic guanosine monophosphate (cGMP) pathway [35].
Tumor cells producing PGE2 can promote MDSCs immunosuppression by activating TGFβ through PGE2 receptors Ep2 and Ep4 found in monocytes [125].
Tumor cells producing PGE2 can promote MDSCs immunosuppression by activating TGFβ through PGE2 receptors Ep2 and Ep4 found in monocytes [36].
In a concluded clinical trial, the oral administration of CSF-1R inhibitor PLX3397 was well tolerated but not efficient to treat GBM patients [129]. These studies have shown partial efficacy of CSF-1R inhibition and the potential benefit of combining it with other therapies to overcome resistance.
In a concluded clinical trial, the oral administration of CSF-1R inhibitor PLX3397 was well tolerated but not efficient to treat GBM patients [37]. These studies have shown partial efficacy of CSF-1R inhibition and the potential benefit of combining it with other therapies to overcome resistance.
The use of CCR2 antagonist CCX872 reduced intratumoral MDSCs and enhanced the anti-PD-1 treatment in GBM-bearing mice [130].
The use of CCR2 antagonist CCX872 reduced intratumoral MDSCs and enhanced the anti-PD-1 treatment in GBM-bearing mice [38].
Low levels of chemotherapies can be used to deplete MDSCs.
Other therapeutic approaches are being developed to promote MDSCs differentiation dampening their pro-tumor activity. MDSCs isolated from cancer patients were differentiated into dendritic cells (DC) using ATRA and GM-CSF (Al). In MDSCs, ATRA causes upregulation of glutathione synthase resulting in the accumulation of glutathione and reduction of ROS [135]. In brain cancer cells (glioma and medulloblastoma), ATRA treatments cause cell growth arrest [136,137,138]. However, the combination of ATRA with epigenetic drugs SAHA and 5-AZA exacerbated tumor growth in a glioma xenograft mouse model [139]. The effects of ATRA in MDSCs of brain tumors remain to be determined.
Other therapeutic approaches are being developed to promote MDSCs differentiation dampening their pro-tumor activity. MDSCs isolated from cancer patients were differentiated into dendritic cells (DC) using ATRA and GM-CSF (Al). In MDSCs, ATRA causes upregulation of glutathione synthase resulting in the accumulation of glutathione and reduction of ROS [39]. In brain cancer cells (glioma and medulloblastoma), ATRA treatments cause cell growth arrest [40][41][42]. However, the combination of ATRA with epigenetic drugs SAHA and 5-AZA exacerbated tumor growth in a glioma xenograft mouse model [43]. The effects of ATRA in MDSCs of brain tumors remain to be determined.
8. Conclusions, Open Questions, and Future Perspectives
So far clinical approaches aimed at targeting MDSCs in human cancer patients have not lived up to the expectations. The reasons for this are due to the incomplete understanding of MDSC phenotypic and functional heterogeneity and the strategies utilized by MDSCs to resist chemo, radiation, and immunotherapies. Some of this can be addressed in preclinical tumor models using state-of-the-art technologies such as single-cell RNA-sequencing (scRNA-seq), nanostring, mass cytometry, multiplexed immunohistochemistry, single-cell assay for transposase accessible chromatin using sequencing (scATAC-seq), and whole-genome sequencing.
Several outstanding questions remain: First, what are the true phenotypic markers that separate MDSC subsets from monocytes and neutrophils. Second, what are the molecular mechanisms that govern the generation, recruitment, and accumulation of MDSCs in HGG? Third, what metabolic programs are altered in MDSCs? Are these alterations common or different in M-MDSCs compared to PMN-MDSCs? Finally, whether targeting MDSCs using different approaches (as mentioned in Strategies for therapeutic targeting of MDSCs) alone as monotherapy is sufficient for inducing significant and long-term durable responses in the clinic or a combination with other treatments is needed? If combination therapy is required then the details regarding the type, sequence, and timing of these treatments have to be worked out. Finding the answer to these questions in the upcoming years will result in novel and efficient treatment options that would improve the survival of high-grade glioma patients by precisely targeting MDSCs.
References
- Fu, W.; Wang, W.; Li, H.; Jiao, Y.; Huo, R.; Yan, Z.; Wang, J.; Wang, S.; Wang, J.; Chen, D.; et al. Single-Cell Atlas Reveals Complexity of the Immunosuppressive Microenvironment of Initial and Recurrent Glioblastoma. Front. Immunol. 2020, 11, 835.
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased production of immature myeloid cells in cancer patients: A mechanism of immunosuppression in cancer. J. Immunol. 2001, 166, 678–689.
- Kamran, N.; Kadiyala, P.; Saxena, M.; Candolfi, M.; Li, Y.; Moreno-Ayala, M.A.; Raja, N.; Shah, D.; Lowenstein, P.R.; Castro, M.G. Immunosuppressive Myeloid Cells’ Blockade in the Glioma Microenvironment Enhances the Efficacy of Immune-Stimulatory Gene Therapy. Mol. Ther. 2017, 25, 232–248.
- Gabrusiewicz, K.; Rodriguez, B.; Wei, J.; Hashimoto, Y.; Healy, L.M.; Maiti, S.N.; Thomas, G.; Zhou, S.; Wang, Q.; Elakkad, A.; et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI Insight 2016, 1, e85841.
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174.
- Yamauchi, Y.; Safi, S.; Blattner, C.; Rathinasamy, A.; Umansky, L.; Juenger, S.; Warth, A.; Eichhorn, M.; Muley, T.; Herth, F.J.F.; et al. Circulating and Tumor Myeloid-derived Suppressor Cells in Resectable Non-Small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2018, 198, 777–787.
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220.
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5, e6017.
- Millrud, C.R.; Bergenfelz, C.; Leandersson, K. On the origin of myeloid-derived suppressor cells. Oncotarget 2017, 8, 3649–3665.
- Solito, S.; Falisi, E.; Diaz-Montero, C.M.; Doni, A.; Pinton, L.; Rosato, A.; Francescato, S.; Basso, G.; Zanovello, P.; Onicescu, G.; et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood 2011, 118, 2254–2265.
- Heine, A.; Held, S.A.E.; Schulte-Schrepping, J.; Wolff, J.F.A.; Klee, K.; Ulas, T.; Schmacke, N.A.; Daecke, S.N.; Riethausen, K.; Schultze, J.L.; et al. Generation and functional characterization of MDSC-like cells. Oncoimmunology 2017, 6, e1295203.
- Kohanbash, G.; Okada, H. Myeloid-derived suppressor cells (MDSCs) in gliomas and glioma-development. Immunol. Investig. 2012, 41, 658–679.
- Zhu, X.; Fujita, M.; Snyder, L.A.; Okada, H. Systemic delivery of neutralizing antibody targeting CCL2 for glioma therapy. J. Neurooncol. 2011, 104, 83–92.
- Alban, T.J.; Alvarado, A.G.; Sorensen, M.D.; Bayik, D.; Volovetz, J.; Serbinowski, E.; Mulkearns-Hubert, E.E.; Sinyuk, M.; Hale, J.S.; Onzi, G.R.; et al. Global immune fingerprinting in glioblastoma patient peripheral blood reveals immune-suppression signatures associated with prognosis. JCI Insight 2018, 3.
- Moertel, C.L.; Xia, J.; LaRue, R.; Waldron, N.N.; Andersen, B.M.; Prins, R.M.; Okada, H.; Donson, A.M.; Foreman, N.K.; Hunt, M.A.; et al. CD200 in CNS tumor-induced immunosuppression: The role for CD200 pathway blockade in targeted immunotherapy. J. Immunother. Cancer 2014, 2, 46.
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Gyorffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660.
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682.
- Alban, T.J.; Bayik, D.; Otvos, B.; Rabljenovic, A.; Leng, L.; Jia-Shiun, L.; Roversi, G.; Lauko, A.; Momin, A.A.; Mohammadi, A.M.; et al. Glioblastoma Myeloid-Derived Suppressor Cell Subsets Express Differential Macrophage Migration Inhibitory Factor Receptor Profiles That Can Be Targeted to Reduce Immune Suppression. Front. Immunol. 2020, 11, 1191.
- Chiu, D.K.C.; Tse, A.P.W.; Xu, I.M.J.; Di Cui, J.; Lai, R.K.H.; Li, L.L.; Koh, H.Y.; Tsang, F.H.C.; Wei, L.L.; Wong, C.M.; et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat. Commun. 2017, 8, 1–12.
- Chiu, D.K.C.; Xu, I.M.J.; Lai, R.K.H.; Tse, A.P.W.; Wei, L.L.; Koh, H.Y.; Li, L.L.; Lee, D.; Lo, R.C.L.; Wong, C.M.; et al. Hypoxia induces myeloid-derived suppressor cell recruitment to hepatocellular carcinoma through chemokine (C-C motif) ligand 26. Hepatology 2016, 64, 797–813.
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453.
- De Leo, A.; Ugolini, A.; Veglia, F. Myeloid Cells in Glioblastoma Microenvironment. Cells 2020, 10, 18.
- Yan, D.; Adeshakin, A.O.; Xu, M.; Afolabi, L.O.; Zhang, G.; Chen, Y.H.; Wan, X. Lipid Metabolic Pathways Confer the Immunosuppressive Function of Myeloid-Derived Suppressor Cells in Tumor. Front. Immunol. 2019, 10, 1399.
- Raychaudhuri, B.; Rayman, P.; Ireland, J.; Ko, J.; Rini, B.; Borden, E.C.; Garcia, J.; Vogelbaum, M.A.; Finke, J. Myeloid-derived suppressor cell accumulation and function in patients with newly diagnosed glioblastoma. Neuro. Oncol. 2011, 13, 591–599.
- Gielen, P.R.; Schulte, B.M.; Kers-Rebel, E.D.; Verrijp, K.; Bossman, S.A.; Ter Laan, M.; Wesseling, P.; Adema, G.J. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro. Oncol. 2016, 18, 1253–1264.
- Lindau, D.; Gielen, P.; Kroesen, M.; Wesseling, P.; Adema, G.J. The immunosuppressive tumour network: Myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 2013, 138, 105–115.
- Wan, Y.Y. Regulatory T cells: Immune suppression and beyond. Cell Mol. Immunol. 2010, 7, 204–210.
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta 1. J. Immunol. 2009, 182, 240–249.
- Tao, J.; Han, D.; Gao, S.; Zhang, W.; Yu, H.; Liu, P.; Fu, R.; Li, L.; Shao, Z. CD8(+) T cells exhaustion induced by myeloid-derived suppressor cells in myelodysplastic syndromes patients might be through TIM3/Gal-9 pathway. J. Cell Mol. Med. 2020, 24, 1046–1058.
- Hou, A.; Hou, K.; Huang, Q.; Lei, Y.; Chen, W. Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 783.
- Clavijo, P.E.; Moore, E.C.; Chen, J.; Davis, R.J.; Friedman, J.; Kim, Y.; Van Waes, C.; Chen, Z.; Allen, C.T. Resistance to CTLA-4 checkpoint inhibition reversed through selective elimination of granulocytic myeloid cells. Oncotarget 2017, 8, 55804–55820.
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310.
- Ramachandran, C.; Nair, S.M.; Escalon, E.; Melnick, S.J. Potentiation of etoposide and temozolomide cytotoxicity by curcumin and turmeric force™ in brain tumor cell lines. J. Complement Integr. Med. 2012, 9.
- Gersey, Z.C.; Rodriguez, G.A.; Barbarite, E.; Sanchez, A.; Walters, W.M.; Ohaeto, K.C.; Komotar, R.J.; Graham, R.M. Curcumin decreases malignant characteristics of glioblastoma stem cells via induction of reactive oxygen species. BMC Cancer 2017, 17, 99.
- Peak, T.C.; Richman, A.; Gur, S.; Yafi, F.A.; Hellstrom, W.J. The Role of PDE5 Inhibitors and the NO/cGMP Pathway in Cancer. Sex. Med. Rev. 2016, 4, 74–84.
- Mao, Y.; Sarhan, D.; Steven, A.; Seliger, B.; Kiessling, R.; Lundqvist, A. Inhibition of tumor-derived prostaglandin-e2 blocks the induction of myeloid-derived suppressor cells and recovers natural killer cell activity. Clin. Cancer Res. 2014, 20, 4096–4106.
- Butowski, N.; Colman, H.; De Groot, J.F.; Omuro, A.M.; Nayak, L.; Wen, P.Y.; Cloughesy, T.F.; Marimuthu, A.; Haidar, S.; Perry, A.; et al. Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: An Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro. Oncol. 2016, 18, 557–564.
- Flores-Toro, J.A.; Luo, D.; Gopinath, A.; Sarkisian, M.R.; Campbell, J.J.; Charo, I.F.; Singh, R.; Schall, T.J.; Datta, M.; Jain, R.K.; et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proc. Natl. Acad. Sci. USA 2020, 117, 1129–1138.
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 2007, 67, 11021–11028.
- Wang, R.; Liu, C. All-trans retinoic acid therapy induces asymmetric division of glioma stem cells from the U87MG cell line. Oncol. Lett. 2019, 18, 3646–3654.
- Chang, Q.; Chen, Z.; You, J.; McNutt, M.A.; Zhang, T.; Han, Z.; Zhang, X.; Gong, E.; Gu, J. All-trans-retinoic acid induces cell growth arrest in a human medulloblastoma cell line. J. Neurooncol. 2007, 84, 263–267.
- Gumireddy, K.; Sutton, L.N.; Phillips, P.C.; Reddy, C.D. All-trans-retinoic acid-induced apoptosis in human medulloblastoma: Activation of caspase-3/poly(ADP-ribose) polymerase 1 pathway. Clin. Cancer Res. 2003, 9, 4052–4059.
- Schmoch, T.; Gal, Z.; Mock, A.; Mossemann, J.; Lahrmann, B.; Grabe, N.; Schmezer, P.; Lasitschka, F.; Beckhove, P.; Unterberg, A.; et al. Combined Treatment of ATRA with Epigenetic Drugs Increases Aggressiveness of Glioma Xenografts. Anticancer Res. 2016, 36, 1489–1496.