Itch or pruritus is the hallmark of atopic dermatitis and is defined as an unpleasant sensation that evokes the desire to scratch. It is also believed that itch is a signal of danger from various environmental factors or physiological abnormalities. Because histamine is a well-known substance inducing itch, H1-antihistamines are the most frequently used drugs to treat pruritus.
- atopic dermatitis
- dry skin
- intractable itch
- itch/pruritus
- keratinocyte
- sensory nerve fiber
1. Introduction
Patients with atopic dermatitis (AD) suffer from recurrent dermatitis and intractable itch. The most frequent clinical phenotype of AD is lichenified/exudative flexural dermatitis alone or associated with head/neck eczema or hand eczema [1]. Although there is no mortality directly associated with AD, this condition substantially impacts patients’ quality of life. In addition to the social stigmatization due to visible skin lesions, severe pruritus can disrupt sleep in patients with AD, which can lead to psychosocial comorbidities, including depression, anxiety, and suicidal ideations [2]. The economic burden of AD is attributed to not only direct medical costs but also to costs through lost work productivity. For instance, in Europe, the direct annual costs have been estimated to EUR 7000, while the indirect costs ranged from EUR 7000 to EUR 14,000 per patient with severe AD [2]. Taken together, patients with AD and society will benefit from the reduction of the burden caused by economic and psychosocial comorbidities in AD.
Although the pathogenesis of AD has not been fully elucidated, it is influenced in a complicated manner by genetic factors, immune dysfunction, physical conditions, stress and weather [3,4,5][3][4][5]. Furthermore, the observation that AD prevalence appears to be increasing in developing countries suggests an important role of environmental factors in the pathogenic mechanism of AD [6]. Because histamine is a well-known pruritogen that is involved in itch accompanied by urticaria caused by the degranulation of mast cells, H1-antihistamines are the most frequently used drugs to treat pruritus. However, H1-antihistamines, even at high doses, are not fully effective against intractable itch in patients with AD [7]. Since severe pruritus leads to disturbances in patient sleep and work that markedly decrease quality of life, H1-antihistamine-resistant itch is a clinical problem in AD. Thus, understanding the pathogenesis of itch and its treatment is important.
2. Transduction of Itch
It is believed that itch is a signal of danger from various environmental factors or physiological abnormalities. Itch initially originates from pruritogens, which are caused by inflammation, dryness or other skin damage. Pruritogens activate certain receptors on the free nerve endings of sensory neurons. Itch sensation is mediated by primary (peripheral) sensory afferents, especially C-fibers, which have cell bodies in the dorsal root ganglia or trigeminal ganglia [8,9][8][9]. Pruritogen receptors are mostly G protein-coupled receptors (GPCRs) that promote the opening of the ion channel group transient receptor potential cation channels, especially transient receptor potential vanilloid 1 (TRPV1) and transient receptor potential ankyrin 1 (TRPA1) [10]. TRPV1 and TRPA1 are activated by diverse stimuli in addition to GPCR ligands, including extracellular pH, adenosine triphosphate (ATP), prostaglandins, oxidants, capsaicin, allyl isothiocyanate, heat and cold, and TRPV1 and TRPA1, which are involved in the pathology of AD and pruritus [11,12][11][12]. The pruritus signal of peripheral sensory afferents is transmitted through the spinal cord in the dorsal root ganglia or trigeminal ganglia to the somatosensory cortex, resulting in recognition of an itch sensation.
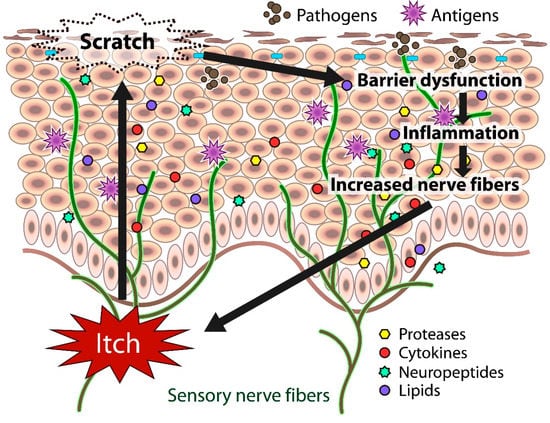
In atopic skin, increased penetration of pathogens and antigens and nerve fiber density with pruritogen receptors elicit severe pruritus following the skin barrier dysfunction or inflammation (Figure 1). In addition, the reduced itch threshold of atopic skin compared with that of healthy skin provokes an abnormal itch sensation called “alloknesis” and “hyperknesis”. Alloknesis is a pathology in which pruritus is elicited by innocuous mechanical stimulation, including contact with fabrics, dressing and undressing [13]. In contrast, hyperknesis represents increased itch elicited by pruritogens, and the sensation of pruritus is stronger in patients with AD than in healthy individuals. Moreover, noxious stimuli evoke pruritus instead of pain in AD patients [14]. Therefore, in AD, itch sensation evokes the desire to scratch, and scratching induces the production of inflammatory cytokines by keratinocytes, further exacerbating eczema and inflammation in the skin (itch–scratch cycle) (Figure 2) [5,13][5][13].
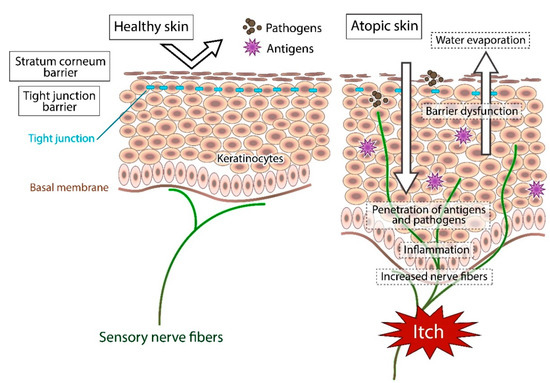
In healthy skin, the penetration of pathogens or antigens and excessive water evaporation are prevented by the stratum corneum barrier and tight junction barrier. Furthermore, the cutaneous nerve fibers terminate in the dermis. In atopic skin, the transepidermal water loss (water evaporation) is increased and the damaged skin barrier enhances penetration of pathogens and antigens. Furthermore, recurrent dermatitis is accompanied by intractable itch, which is induced by inflammation and epidermal hyperinnervation.
Itch sensation evokes the desire to scratch, and scratching impairs skin barrier function. The barrier dysfunction leads to the production of inflammatory and itch mediators (cytokines, proteases, neuropeptides, lipids) by cutaneous cells, further exacerbating eczema and inflammation in the skin. Both barrier dysfunction and inflammation contribute to the increased number of fibers.
3. Substances and Receptors Inducing Itch in AD
3.1. Histamine
Histamine is involved primarily in acute itch and is a cause of itch in urticaria and insect bite reactions. Histamine is produced mainly by mast cells, although other cells, including basophils, keratinocytes and neurons in the skin, can also release it [15]. Although four receptors have been identified as histamine receptors, H1, H2, H3 and H4, only the H1 and H4 receptors activate TRPV1 and are considered therapeutic targets for pruritus [16]. However, H1-antihistamines do not improve intractable itch in patients with AD, implying that chronic itch is largely induced by a histamine-independent pathway [7,8][7][8]. The application of antihistamines to AD model mice is also known to be ineffective. In NC/Nga mice (which spontaneously develop AD-like skin lesions when they are raised in conventional circumstances), long-duration scratching (longer than one second) is not suppressed by the H1 receptor antagonist chlorpheniramine or the H2 receptor antagonist famotidine and the H3/4 receptor antagonist thioperamide [17]. Additionally, treatment with the H4 receptor antagonist JNJ7777120 or JNJ28307474 does not affect the inhibition of scratching behavior or amelioration of AD in NC/Nga mice [18].
3.2. Proteases
Several proteases produced by cutaneous cells or exogenous biotic factors, including bacteria, mites and plants, are involved in pruritus in AD. Endogenous proteases include tryptases, chymases, trypsins, and kallikreins and are produced by keratinocytes, mast cells, macrophages, dendritic cells, B cells, T cells, and neutrophils [19]. The number of tryptase-positive mast cells is increased in the upper dermis of the lesional and nonlesional skin of patients with AD [20]. In addition, various kallikrein proteases are upregulated in the stratum corneum of patients with AD [21].
Proteases cleave the extracellular domain of protease-activated receptors (PARs), and then the new amino terminus of the receptor itself acts as a PAR ligand. To date, four PARs, PAR-1, PAR-2, PAR-3, and PAR-4, have been identified. PAR-1, PAR-2 and PAR-4 are involved in acute itch in mice activated by the specific peptides TFLLR, SLIGRL, and AYPGKF, respectively [22]. PAR-1, PAR-2, and PAR-4 are expressed in cutaneous nerve fibers, keratinocytes, mast cells, and macrophages [19]. The number of PAR-2-positive keratinocytes and the activity of serine proteases are increased in the skin of AD model NC mice [23]. Plant cysteine proteases, including mucunain (cowhage), bromelain (pineapple stem), ficin (fig tree latex), and papain (papaya), induce itch via activation of PAR-2 and PAR-4 [24,25][24][25].
H1-antihistamines inhibit itch in mice elicited by agonists of PAR-1 and PAR-4 but not agonists of PAR-2, suggesting the involvement of PAR-2 in histamine-independent itch [22]. Given that scratching evoked by trypsin, which acts on PAR-1, PAR-2, and PAR-4, is suppressed by TRPV1 inhibition or TRPV1 knockout in mice, as well as by the H1-antihistamine cyproheptadine, trypsin-evoked pruritus is considered to be involved in histamine-dependent itch [26]. In PAR-2 knockout mice, trypsin- and SLIGRL-evoked scratching still occur; however, tryptase-evoked scratching is suppressed in these mice and is also inhibited by an anti-PAR-2 neutralizing antibody and the PAR-2 antagonist FSLLRY [27]. These findings suggest that different signaling pathways are activated by different agonists and/or that the roles of PAR-2 in neurons and keratinocytes differ. The serine protease inhibitor nafamostat mesilate inhibits tryptase-evoked scratching and spontaneous scratching in AD model NC mice [23,28][23][28]. An anti-PAR-2 antibody was also shown to suppress spontaneous scratching in NC mice [23]. Topical application of tacrolimus (the calcineurin inhibitor FK506) inhibits SLIGRL-evoked scratching, suggesting that its antipruritic effect is due to PAR-2 signaling inhibition [29].
3.3. Cytokines
Some cytokines are responsible not only for inflammation but also for itch sensation. Interleukin (IL)-31 is a cytokine mainly produced by type 2 helper T cells (Th2 cells) that exhibits upregulated expression in the lesional skin of patients with skin disease accompanied by itch, including AD, contact dermatitis and prurigo nodularis [30,31,32][30][31][32]. In addition to Th2 cells, mast cells, macrophages, dendritic cells, and eosinophils also produce IL-31 [33]. It has been reported that IL-31 participates in the itch sensation and promotes long-lasting scratching behavior in NC/Nga and BALB/c mice [34]. The IL-31 receptor is a heterodimer that consists of IL-31 receptor A and an oncostatin M receptor. IL-31 evokes itch directly via IL-31 receptor A expressed in sensory neurons via both TRPV1 and TRPA1 activation [35]. A missense mutation in the oncostatin M-specific receptor subunit β gene was found in families affected by familial primary localized cutaneous amyloidosis, an autosomal-dominant skin disease associated with chronic, severe pruritus [36]. It has been reported that the humanized anti-human IL-31 receptor A monoclonal antibody CIM331 (nemolizumab) reduces the severity and pruritus score of AD [37,38][37][38].
A second cytokine contributing to itch is thymic stromal lymphopoietin (TSLP), which is secreted primarily by keratinocytes, mast cells, and dendritic cells and promotes Th2 immune responses [39,40][39][40]. The expression of TSLP is upregulated in the epidermal keratinocytes of AD patients [41,42][41][42]. TSLP binds a heterodimeric receptor comprising the IL-7 receptor α chain and TSLP receptor expressed in dendritic cells, T cells, B cells, mast cells, basophils, and eosinophils [43,44][43][44]. TSLP directly elicits itch via the TSLP receptor on TRPA1-positive sensory neurons [42]. Interestingly, in return, scratching induces the production of TSLP by keratinocytes, leading to an itch–scratch cycle [45].
Both IL-4 and IL-13 are well-known Th2 cytokines overexpressed in AD skin. It has been shown that IL-4 and IL-13 are involved in chronic but not acute itch by directly activating sensory neurons via the IL-4 receptor α chain [46]. The IL-4 receptor α (IL-4Ra) chain is shared by IL-4 and IL-13 receptors and activates janus kinase (JAK) 1, suggesting that blockade of IL-4Ra or JAK1 signaling in sensory neurons might be a useful treatment for chronic itch. In fact, the fully human monoclonal antibody dupilumab, which blocks binding to IL-4Ra, reduces the severity and pruritus score of AD [47,48][47][48]. Moreover, baricitinib, a selective JAK1 and JAK2 inhibitor, reduces pruritus and inflammation in patients with moderate to severe AD [49]. In addition, IL-13 antibodies, lebrikizumab and tralokinumab, improve AD symptoms although they do not show strong effect on pruritus [50,51][50][51]. Overall, more biologic drugs blocking IL-4 and IL-13 are under development and will be available in the future for AD treatment [52,53,54][52][53][54].
3.4. Neuropeptides
Neuropeptides play key roles in modulating neuronal activity. Substance P is a tachykinin neuropeptide that is involved in AD itch [55]. Substance P is secreted by sensory neurons and keratinocytes and elicits itch by histamine-dependent and histamine-independent mechanisms. A relatively high density of substance-P-containing nerve fibers has been observed in AD skin [56]. Neurokinin 1 receptor (NK1R) is a substance P receptor involved in itch and is expressed in sensory neurons, mast cells, keratinocytes, and fibroblasts. These cells release additional mediators inducing pruritus following stimulation with substance P. Although some patients did not respond, a study showed that the oral neurokinin 1 receptor antagonist aprepitant demonstrated efficacy in the treatment of intractable pruritus in AD and prurigo nodularis [57].
Substance P and calcitonin-gene-related peptide (CGRP) have roles in itch sensation hypersensitivity and neurogenic inflammation [58]. The release of both substance P and CGRP from sensory neurons excited by histamine leads to local vasodilation, plasma extravasation, and mast cell degranulation, whose response is neurogenic inflammation [59,60][59][60]. These findings indicate that neuropeptides play important roles in chronic itch under the pathological conditions of AD.
3.5. Lipids
Lipid mediators are produced by various pathophysiological stimuli that contribute to the pathogenesis of various inflammatory skin diseases. The roles of lipid mediators in the pathogenesis of AD and intractable pruritus remain largely unknown. Arachidonic acid released from membrane phospholipids is converted by each synthase into lipid mediators, including prostanoids, leukotrienes (LTs), and hydroxy-eicosatetraenoic acids [61]. Prostanoids, encompassing prostaglandin (PG) and thromboxane (TX), are synthesized by cyclooxygenase. Finally, arachidonic acid is metabolized to PGD2, PGE2, PGF2α, PGI2, and TXA2 by their specific synthesis pathways. Prostanoids are released from cells immediately after synthesis and are chemically and metabolically unstable. These lipids thereby act on target cells only locally via GPCRs [61]. The concentrations of PGE2 and LTB4 are elevated in the lesional skin of patients with AD, suggesting that these mediators are involved in biochemical processes leading to AD through cutaneous inflammation [62]. It has been demonstrated that PGE2 acts as a weak pruritogen and potent vasodilator in normal skin as well as in the skin of patients with AD without induction of protein extravasation [63]. Furthermore, an antagonist of LTB4 receptor, ONO-4057, has been shown to inhibit spontaneous scratching of NC mice with chronic dermatitis [64]. A stable analog of TXA2, U-46619, has been shown to elicit itch in mice through thromboxane prostanoid receptors expressed in cutaneous nerve fibers and keratinocytes [65]. This TXA2-induced scratching behavior is inhibited by the thromboxane prostanoid receptor antagonist ONO-3708 and thromboxane prostanoid receptor deficiency in mice [65].
On the other hand, topical application of PGD2, PGI1, PGE1, PGE2 or arachidonic acid was shown to suppress scratching in NC/Nga mice with AD, while the cyclooxygenase inhibitor indomethacin enhanced scratching [66]. Among the above lipid mediators, PGD2 is the most effective mediator, and its inhibition depends on the prostanoid DP1 receptor but not on the DP2 receptor, indicating the therapeutic potential of prostanoid DP1 receptor agonists [66].
3.6. Opioids
Opioids are peptides that have pharmacological effects similar to those of morphine. The endogenous opioids β-endorphin and dynorphins activate the µ-opioid receptor and κ-opioid receptor, and the balance of this activation plays pivotal roles in the regulation of pruritus in both the central and peripheral nervous systems [67]. Opioid receptors are GPCRs that also recognize exogenous opioids, such as opiates and alkaloids. It is thought that opioid receptors can contribute to cell differentiation, migration, wound healing, and immunity in human skin [68]. The expression of µ-opioid receptor has been observed in the epidermal keratinocytes and primary sensory afferents of normal skin [69,70][69][70]. The serum concentration of β-endorphin is increased in patients with AD and is correlated with both itch intensity and disease severity [71]. Opioid-induced itch is a well-known side effect of pain treatment with morphine, a µ-opioid receptor agonist. In contrast, the µ-opioid receptor antagonist naloxone and κ-opioid receptor agonist nalfurafine decrease pruritus in patients with AD, chronic renal failure or cholestasis [68]. Furthermore, topical application of the µ-opioid receptor antagonist naltrexone also inhibits pruritus in patients with AD [72]. Phototherapy, such as ultraviolet A (UVA) treatment, UVA1 treatment, narrow band (NB)-UVB treatment and excimer lasers or lamps, improves pruritus and dermatitis [73,74][73][74]. It has been reported that dynorphin levels are downregulated in the epidermis of AD patients and that psoralen-ultraviolet A (PUVA) therapy rescues the downregulation and visual analog scale (VAS) scores [75]. Thus, some studies suggest that opioids may be directly associated with modulation of itch.
References
- Nettis, E.; Ortoncelli, M.; Pellacani, G.; Foti, C.; Di Leo, E.; Patruno, C.; Rongioletti, F.; Argenziano, G.; Ferrucci, S.M.; Macchia, L.; et al. A multicenter study on the prevalence of clinical patterns and clinical phenotypes in adult atopic dermatitis. J. Investig. Allergol. Clin. Immunol. 2020, 30, 448–450.
- Girolomoni, G.; Luger, T.; Nosbaum, A.; Gruben, D.; Romero, W.; Llamado, L.J.; DiBonaventura, M. The economic and psychosocial comorbidity burden among adults with moderate-to-severe atopic dermatitis in Europe: Analysis of a cross-sectional survey. Dermatol. Ther. 2021, 11, 117–130.
- David Boothe, W.; Tarbox, J.A.; Tarbox, M.B. Atopic Dermatitis: Pathophysiology. Adv. Exp. Med. Biol. 2017, 1027, 21–37.
- Murota, H.; Katayama, I. Exacerbating factors of itch in atopic dermatitis. Allergol. Int. 2017, 66, 8–13.
- Suárez, A.L.; Feramisco, J.D.; Koo, J.; Steinhoff, M. Psychoneuroimmunology of psychological stress and atopic dermatitis: Pathophysiologic and therapeutic updates. Acta Derm. Venereol. 2012, 92, 7–15.
- Bonamonte, D.; Filoni, A.; Vestita, M.; Romita, P.; Foti, C.; Angelini, G. The role of the environmental risk factors in the pathogenesis and clinical outcome of atopic dermatitis. Biomed. Res. Int. 2019, 2019, 2450605.
- Klein, P.A.; Clark, R.A.F. An evidence-based review of the efficacy of antihistamines in relieving pruritus in atopic dermatitis. Arch. Dermatol. 1999, 135, 1522–1525.
- Ikoma, A.; Steinhoff, M.; Ständer, S.; Yosipovitch, G.; Schmelz, M. The neurobiology of itch. Nat. Rev. Neurosci. 2006, 7, 535–547.
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284.
- Kittaka, H.; Tominaga, M. The molecular and cellular mechanisms of itch and the involvement of TRP channels in the peripheral sensory nervous system and skin. Allergol. Int. 2017, 66, 22–30.
- Tóth, B.I.; Oláh, A.; Szöllősi, A.G.; Bíró, T. TRP channels in the skin. Br. J. Pharmacol. 2014, 171, 2568–2581.
- Sousa-Valente, J.; Andreou, A.P.; Urban, L.; Nagy, I. Transient receptor potential ion channels in primary sensory neurons as targets for novel analgesics. Brit. J. Pharmacol. 2014, 171, 2508–2527.
- Yosipovitch, G.; Greaves, M.W.; Schmelz, M. Itch. Lancet 2003, 361, 690–694.
- Ikoma, A.; Fartasch, M.; Heyer, G.; Miyachi, Y.; Handwerker, H.; Schmelz, M. Painful stimuli evoke itch in patients with chronic pruritus: Central sensitization for itch. Neurology 2004, 62, 212–217.
- Thurmond, R.L.; Kazerouni, K.; Chaplan, S.R.; Greenspan, A.J. Peripheral Neuronal Mechanism of Itch: Histamine and Itch. In Itch: Mechanisms and Treatment; Carstens, E., Akiyama, T., Eds.; CRC Press: Boca Raton, FL, USA, 2014.
- Ohsawa, Y.; Hirasawa, N. The role of histamine H1 and H4 receptors in atopic dermatitis: From basic research to clinical study. Allergol. Int. 2014, 63, 533–542.
- Hashimoto, Y.; Takano, N.; Nakamura, A.; Nakaike, S.; Yu, Z.; Endo, Y.; Arai, I. Scratching behavior in NC/Nga mice with dermatitis: Involvement of histamine-induced itching. Allergol. Int. 2004, 53, 349–358.
- Kamo, A.; Negi, O.; Tengara, S.; Kamata, Y.; Noguchi, A.; Ogawa, H.; Tominaga, M.; Takamori, K. Histamine H(4) receptor antagonists ineffective against itch and skin inflammation in atopic dermatitis mouse model. J. Investig. Dermatol. 2014, 134, 546–548.
- Akiyama, T.; Lerner, E.A.; Carstens, E. Protease-Activated Receptors and Itch. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2015; Volume 226, pp. 219–235.
- Järvikallio, A.; Naukkarinen, A.; Harvima, I.T.; Aalto, M.L.; Horsmanheimo, M. Quantitative analysis of tryptase- and chymase-containing mast cells in atopic dermatitis and nummular eczema. Br. J. Dermatol. 1997, 136, 871–877.
- Komatsu, N.; Saijoh, K.; Kuk, C.; Liu, A.C.; Khan, S.; Shirasaki, F.; Takehara, K.; Diamandis, E.P. Human tissue kallikrein expression in the stratum corneum and serum of atopic dermatitis patients. Exp. Dermatol. 2007, 16, 513–519.
- Tsujii, K.; Andoh, T.; Lee, J.B.; Kuraishi, Y. Activation of proteinase-activated receptors induces itch-associated response through histamine-dependent and -independent pathways in mice. J. Pharmacol. Sci. 2008, 108, 385–388.
- Tsujii, K.; Andoh, T.; Ui, H.; Lee, J.B.; Kuraishi, Y. Involvement of tryptase and proteinase-activated receptor-2 in spontaneous itch-associated response in mice with atopy-like dermatitis. J. Pharmacol. Sci. 2009, 109, 388–395.
- Reddy, V.B.; Iuga, A.O.; Shimada, S.G.; LaMotte, R.H.; Lerner, E.A. Cowhage-evoked itch is mediated by a novel cysteine protease: A ligand of protease-activated receptors. J. Neurosci. 2008, 28, 4331–4335.
- Reddy, V.B.; Lerner, E.A. Plant cysteine proteases that evoke itch activate protease-activated receptors. Br. J. Dermatol. 2010, 163, 532–535.
- Costa, R.; Marotta, D.M.; Manjavachi, M.N.; Fernandes, E.S.; Lima-Garcia, J.F.; Paszcuk, A.F.; Quintao, N.L.; Juliano, L.; Brain, S.D.; Calixto, J.B. Evidence for the role of neurogenic inflammation components in trypsin-elicited scratching behaviour in mice. Br. J. Pharmacol. 2008, 154, 1094–1103.
- Liu, Q.; Weng, H.J.; Patel, K.N.; Tang, Z.; Bai, H.; Steinhoff, M.; Dong, X. The distinct roles of two GPCRs, MrgprC11 and PAR2, in itch and hyperalgesia. Sci. Signal. 2011, 4, ra45.
- Ui, H.; Andoh, T.; Lee, J.B.; Nojima, H.; Kuraishi, Y. Potent pruritogenic action of tryptase mediated by PAR-2 receptor and its involvement in anti-pruritic effect of nafamostat mesilate in mice. Eur. J. Pharmacol. 2006, 530, 172–178.
- Nakano, T.; Andoh, T.; Tayama, M.; Kosaka, M.; Lee, J.B.; Kuraishi, Y. Effects of topical application of tacrolimus on acute itch-associated responses in mice. Biol. Pharm. Bull. 2008, 31, 752–754.
- Sonkoly, E.; Muller, A.; Lauerma, A.I.; Pivarcsi, A.; Soto, H.; Kemeny, L.; Alenius, H.; Dieu-Nosjean, M.C.; Meller, S.; Rieker, J.; et al. IL-31: A new link between T cells and pruritus in atopic skin inflammation. J. Allergy Clin. Immunol. 2006, 117, 411–417.
- Neis, M.M.; Peters, B.; Dreuw, A.; Wenzel, J.; Bieber, T.; Mauch, C.; Krieg, T.; Stanzel, S.; Heinrich, P.C.; Merk, H.F.; et al. Enhanced expression levels of IL-31 correlate with IL-4 and IL-13 in atopic and allergic contact dermatitis. J. Allergy Clin. Immunol. 2006, 118, 930–937.
- Raap, U.; Wichmann, K.; Bruder, M.; Ständer, S.; Wedi, B.; Kapp, A.; Werfel, T. Correlation of IL-31 serum levels with severity of atopic dermatitis. J. Allergy Clin. Immunol. 2008, 122, 421–423.
- Gibbs, B.F.; Patsinakidis, N.; Raap, U. Role of the pruritic cytokine IL-31 in autoimmune skin diseases. Front. Immunol. 2019, 10, 1383.
- Arai, I.; Tsuji, M.; Takeda, H.; Akiyama, N.; Saito, S. A single dose of interleukin-31 (IL-31) causes continuous itch-associated scratching behaviour in mice. Exp. Dermatol. 2013, 22, 669–671.
- Cevikbas, F.; Wang, X.; Akiyama, T.; Kempkes, C.; Savinko, T.; Antal, A.; Kukova, G.; Buhl, T.; Ikoma, A.; Buddenkotte, J.; et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: Involvement of TRPV1 and TRPA1. J. Allergy Clin. Immunol. 2014, 133, 448–460.
- Arita, K.; South, A.P.; Hans-Filho, G.; Sakuma, T.H.; Lai-Cheong, J.; Clements, S.; Odashiro, M.; Odashiro, D.N.; Hans-Neto, G.; Hans, N.R.; et al. Oncostatin M receptor-beta mutations underlie familial primary localized cutaneous amyloidosis. Am. J. Hum. Genet. 2008, 82, 73–80.
- Nemoto, O.; Furue, M.; Nakagawa, H.; Shiramoto, M.; Hanada, R.; Matsuki, S.; Imayama, S.; Kato, M.; Hasebe, I.; Taira, K.; et al. The first trial of CIM331, a humanized antihuman interleukin-31 receptor A antibody, in healthy volunteers and patients with atopic dermatitis to evaluate safety, tolerability and pharmacokinetics of a single dose in a randomized, double-blind, placebo-controlled study. Br. J. Dermatol. 2016, 174, 296–304.
- Ruzicka, T.; Hanifin, J.M.; Furue, M.; Pulka, G.; Mlynarczyk, I.; Wollenberg, A.; Galus, R.; Etoh, T.; Mihara, R.; Yoshida, H.; et al. Anti-interleukin-31 receptor A antibody for atopic dermatitis. N. Engl. J. Med. 2017, 376, 826–835.
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680.
- Kashyap, M.; Rochman, Y.; Spolski, R.; Samsel, L.; Leonard, W.J. Thymic stromal lymphopoietin is produced by dendritic cells. J. Immunol. 2011, 187, 1207–1211.
- Moniaga, C.S.; Jeong, S.K.; Egawa, G.; Nakajima, S.; Hara-Chikuma, M.; Jeon, J.E.; Lee, S.H.; Hibino, T.; Miyachi, Y.; Kabashima, K. Protease activity enhances production of thymic stromal lymphopoietin and basophil accumulation in flaky tail mice. Am. J. Pathol. 2013, 182, 841–851.
- Wilson, S.R.; The, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013, 155, 285–295.
- Pandey, A.; Ozaki, K.; Baumann, H.; Levin, S.D.; Puel, A.; Farr, A.G.; Ziegler, S.F.; Leonard, W.J.; Lodish, H.F. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat. Immunol. 2000, 1, 59–64.
- Ziegler, S.F.; Roan, F.; Bell, B.D.; Stoklasek, T.A.; Kitajima, M.; Han, H. The biology of thymic stromal lymphopoietin (TSLP). Adv. Pharmacol. 2013, 66, 129–155.
- Oyoshi, M.K.; Larson, R.P.; Ziegler, S.F.; Geha, R.S. Mechanical injury polarizes skin dendritic cells to elicit a T(H)2 response by inducing cutaneous thymic stromal lymphopoietin expression. J. Allergy Clin. Immunol. 2010, 126, 976–984.e5.
- Oetjen, L.K.; Mack, M.R.; Feng, J.; Whelan, T.M.; Niu, H.; Guo, C.J.; Chen, S.; Trier, A.M.; Xu, A.Z.; Tripathi, S.V.; et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell 2017, 171, 217–228.e13.
- Beck, L.A.; Thaçi, D.; Hamilton, J.D.; Graham, N.M.; Bieber, T.; Rocklin, R.; Ming, J.E.; Ren, H.; Kao, R.; Simpson, E.; et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N. Engl. J. Med. 2014, 371, 130–139.
- Thaçi, D.; Simpson, E.L.; Beck, L.A.; Bieber, T.; Blauvelt, A.; Papp, K.; Soong, W.; Worm, M.; Szepietowski, J.C.; Sofen, H.; et al. Efficacy and safety of dupilumab in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical treatments: A randomised, placebo-controlled, dose-ranging phase 2b trial. Lancet 2016, 387, 40–52.
- Guttman-Yassky, E.; Silverberg, J.I.; Nemoto, O.; Forman, S.B.; Wilke, A.; Prescilla, R.; de la Peña, A.; Nunes, F.P.; Janes, J.; Gamalo, M.; et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: A phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J. Am. Acad. Dermatol. 2019, 80, 913–921.e9.
- Simpson, E.L.; Flohr, C.; Eichenfield, L.F.; Bieber, T.; Sofen, H.; Taïeb, A.; Owen, R.; Putnam, W.; Castro, M.; DeBusk, K.; et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: A randomized, placebo-controlled phase II trial (TREBLE). J. Am. Acad. Dermatol. 2018, 78, 863–871.e11.
- Wollenberg, A.; Howell, M.D.; Guttman-Yassky, E.; Silverberg, J.I.; Kell, C.; Ranade, K.; Moate, R.; van der Merwe, R. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J. Allergy Clin. Immunol. 2019, 143, 135–141.
- Napolitano, M.; Marasca, C.; Fabbrocini, G.; Patruno, C. Adult atopic dermatitis: New and emerging therapies. Expert Rev. Clin. Pharmacol. 2018, 11, 867–878.
- Fabbrocini, G.; Napolitano, M.; Megna, M.; Balato, N.; Patruno, C. Treatment of atopic dermatitis with biologic drugs. Dermatol. Ther. 2018, 8, 527–538.
- Dattola, A.; Bennardo, L.; Silvestri, M.; Nisticò, S.P. What’s new in the treatment of atopic dermatitis? Dermatol. Ther. 2019, 32, e12787.
- Andoh, T.; Kuraishi, Y. Substance P and itch. In Itch: Basic Mechanisms and Therapy; Yosipovitch, G., Ed.; Dekker: New York, NY, USA, 2004; pp. 87–95.
- Järvikallio, A.; Harvima, I.T.; Naukkarinen, A. Mast cells, nerves and neuropeptides in atopic dermatitis and nummular eczema. Arch. Dermatol. Res. 2003, 295, 2–7.
- Ständer, S.; Siepmann, D.; Herrgott, I.; Sunderkötter, C.; Luger, T.A. Targeting the neurokinin receptor 1 with aprepitant: A novel antipruritic strategy. PLoS ONE 2010, 5, e10968.
- Steinhoff, M.; Ständer, S.; Seeliger, S.; Ansel, J.C.; Schmelz, M.; Luger, T. Modern aspects of cutaneous neurogenic inflammation. Arch. Dermatol. 2003, 139, 1479–1488.
- Schmelz, M.; Petersen, L.J. Neurogenic inflammation in human and rodent skin. News Physiol. Sci. 2001, 16, 33–37.
- Rosa, A.C.; Fantozzi, R. The role of histamine in neurogenic inflammation. Br. J. Pharmacol. 2013, 170, 38–45.
- Narumiya, S.; Sugimoto, Y.; Ushikubi, F. Prostanoid receptors: Structures, properties, and functions. Physiol. Rev. 1999, 79, 1193–1226.
- Töröcsik, D.; Weise, C.; Gericke, J.; Szegedi, A.; Lucas, R.; Mihaly, J.; Worm, M.; Rühl, R. Transcriptomic and lipidomic profiling of eicosanoid/docosanoid signalling in affected and non-affected skin of human atopic dermatitis patients. Exp. Dermatol. 2019, 28, 177–189.
- Neisius, U.; Olsson, R.; Rukwied, R.; Lischetzki, G.; Schmelz, M. Prostaglandin E2 induces vasodilation and pruritus, but no protein extravasation in atopic dermatitis and controls. J. Am. Acad. Dermatol. 2002, 47, 28–32.
- Andoh, T.; Haza, S.; Saito, A.; Kuraishi, Y. Involvement of leukotriene B4 in spontaneous itch-related behaviour in NC mice with atopic dermatitis-like skin lesions. Exp. Dermatol. 2011, 20, 894–898.
- Andoh, T.; Nishikawa, Y.; Yamaguchi-Miyamoto, T.; Nojima, H.; Narumiya, S.; Kuraishi, Y. Thromboxane A2 induces itch-associated responses through TP receptors in the skin in mice. J. Investig. Dermatol. 2007, 127, 2042–2047.
- Arai, I.; Takano, N.; Hashimoto, Y.; Futaki, N.; Sugimoto, M.; Takahashi, N.; Inoue, T.; Nakaike, S. Prostanoid DP1 receptor agonist inhibits the pruritic activity in NC/Nga mice with atopic dermatitis. Eur. J. Pharmacol. 2004, 505, 229–235.
- Goldstein, A.; Naidu, A. Multiple opioid receptors: Ligand selectivity profiles and binding site signatures. Mol. Pharmacol. 1989, 36, 265–272.
- Bigliardi, P.L.; Tobin, D.J.; Gaveriaux-Ruff, C.; Bigliardi-Qi, M. Opioids and the skin--where do we stand? Exp. Dermatol. 2009, 18, 424–430.
- Bigliardi-Qi, M.; Bigliardi, P.L.; Eberle, A.N.; Buchner, S.; Rufli, T. beta-endorphin stimulates cytokeratin 16 expression and downregulates mu-opiate receptor expression in human epidermis. J. Investig. Dermatol. 2000, 114, 527–532.
- Ständer, S.; Gunzer, M.; Metze, D.; Luger, T.; Steinhoff, M. Localization of mu-opioid receptor 1A on sensory nerve fibers in human skin. Regul. Pept. 2002, 110, 75–83.
- Lee, C.H.; Chuang, H.Y.; Shih, C.C.; Jong, S.B.; Chang, C.H.; Yu, H.S. Transepidermal water loss, serum IgE and beta-endorphin as important and independent biological markers for development of itch intensity in atopic dermatitis. Br. J. Dermatol. 2006, 154, 1100–1107.
- Bigliardi, P.L.; Stammer, H.; Jost, G.; Rufli, T.; Buchner, S.; Bigliardi-Qi, M. Treatment of pruritus with topically applied opiate receptor antagonist. J. Am. Acad. Dermatol. 2007, 56, 979–988.
- Rivard, J.; Lim, H.W. Ultraviolet phototherapy for pruritus. Dermatol. Ther. 2005, 18, 344–354.
- Legat, F.J. Is there still a role for UV therapy in itch treatment? Exp. Dermatol. 2019, 28, 1432–1438.
- Tominaga, M.; Ogawa, H.; Takamori, K. Possible roles of epidermal opioid systems in pruritus of atopic dermatitis. J. Investig. Dermatol. 2007, 127, 2228–2235.