Human cytomegalovirus (hCMV) is one of the most common causes of congenital infection in the post-rubella era, representing a major public health concern. Although most cases are asymptomatic in the neonatal period, congenital CMV (cCMV) disease can result in permanent impairment of cognitive development and represents the leading cause of non-genetic sensorineural hearing loss. Moreover, even if hCMV mostly causes asymptomatic or pauci-symptomatic infections in immunocompetent hosts, it may lead to severe and life-threatening disease in immunocompromised patients. Since immunity reduces the severity of disease, in the last years, the development of an effective and safe hCMV vaccine has been of great interest to pharmacologic researchers. Both hCMV live vaccines—e.g., live-attenuated, chimeric, viral-based—and non-living ones—subunit, RNA-based, virus-like particles, plasmid-based DNA—have been investigated. Encouraging data are emerging from clinical trials, but a hCMV vaccine has not been licensed yet. Major difficulties in the development of a satisfactory vaccine include hCMV’s capacity to evade the immune response, unclear immune correlates for protection, low number of available animal models, and insufficient general awareness. Moreover, there is a need to determine which may be the best target populations for vaccine administration. The aim of the present paper is to examine the status of hCMV vaccines undergoing clinical trials and understand barriers limiting their development.
- hCMV
- vaccine
- mRNA vaccines
- congenital CMV
- congenital infection
- prevention
- CMV long-term sequelae
- sensorineural hearing loss
1. Epidemiology
Human cytomegalovirus (hCMV), which is one of the eight herpesviruses known to infect humans, is widespread all around the world and is mostly asymptomatic in immunocompetent people [1]. However, hCMV may lead to severe and life-threatening disease in congenitally infected children and immunosuppressed individuals, such as transplant patients and those affected by acquired immunodeficiency syndrome (AIDS) [2].
hCMV is the most frequent cause of vertical transmission infections. The rate of congenital hCMV (cCMV) transmission is between 0.5% and 0.7% of pregnancies in developed nations, and up to 2% of pregnancies in the developing world [3]. The major sources of the virus for expectant mothers are young children with hCMV, most of all toddlers, who can produce saliva and urine with high levels of hCMV [4]. Intrauterine hCMV transmission may occur in mothers without pre-existing antibodies who acquire a primary hCMV infection during pregnancy but also in women who have had previous contacts with hCMV (non-primary infection). In the context of primary infection, 1–4% of seronegative women seroconverted during pregnancy, but the symptoms, when present, are often too mild to seek medical attention. Thus, if hCMV is not tested during pregnancy, the infection is usually not recognized [5][6]. In primary infection, hCMV is transmitted across the placenta in up to 30–50% of the cases, producing fetal infection [5]. Non-primary infection occurs when the fetus is infected because of viral reactivation or in case of maternal reinfection with a different hCMV strain [7]. In such circumstances, the likelihood of hCMV transmission to the fetus is in the range of approximately 3% [5]. However, about three-quarters of cCMV infections are caused by non-primary maternal infection, given the high rates of hCMV seropositivity among women of childbearing age [8]. Vertical transmission is more frequent in mothers with older gestational age, while the risk of fetal damage is higher when infection occurs in the early stages of pregnancy [9][10].
hCMV morbidity and mortality can also occur in immunosuppressed people, mostly AIDS or transplant patients, in whom the virus often behaves like an opportunistic pathogen [11][12]. Before the development of antiretroviral therapy, up to 40% of AIDS patients had severe hCMV disease, often sight-threatening [12], while the introduction of such therapy has led to a rapid decline in the incidence of hCMV manifestations and a better prognosis of patients with hCMV disease [13]. In parallel, hCMV infection represents a frequent complication in the setting of hematopoietic stem cell transplantation (HSCT) and solid organ transplantation (SOT) [11], possibly leading to graft rejection. In the case of SOT, when a hCMV seronegative recipient receives an organ from a seropositive donor, the disease develops in more than 50% of cases if no pre-emptive prophylaxis is given [14]. However, even seropositive recipients may have hCMV disease, given the possibility of superinfection with a new strain or reactivation, even if the latter case is less frequent [15]. Differently, after HSCT, the most frequent cause of disease is hCMV reactivation under the influence of immunosuppression in a seropositive recipient [16]. Antiviral prophylaxis, or treatment when needed, is administered routinely to SOT and HSCT patients to prevent serious disease and is significantly but not completely successful [17]. Finally, hCMV may also pose a danger to intensive care unit (ICU) patients, since it has been demonstrated that it is associated with an increased risk of all-cause mortality, increased hospital and ICU length of stay, longer duration of mechanical ventilation, and increased rates of nosocomial infection [18].
2. Virology
hCMV is a member of the Betaherpesvirinae sub-family of Herpesviridae [19]. It has an icosahedral capsid that contains the double-stranded DNA, encoding for 165 proteins. As shown in
, the capsid is surrounded by a tegument made of proteins and, externally, by a lipid envelope [19], whose glycoproteins allow the entry into the host cells through fusion with the cell membrane. After fusion, the viral capsid with the DNA and the tegument proteins are released into the cell [20]. The virus contains various distinct types of glycoprotein complexes (gC) that are useful to infect the host [21]: gC-I is a complex composed of homotrimers of glycoprotein B (gB), a pan-Herpesviridae conserved glycoprotein, mediator of membrane fusion, that rearranges during entry into the cell from a pre-fusion conformation to a post-fusion one; gC-II is the most abundant gC, is made up of glycoprotein M (gM) and N (gN), contributes to the initial binding to the cell membrane, and plays a role in viral replication; gC-III, now called trimer complex (TC), is a heterotrimeric complex where glycoprotein H (gH), L (gL), and O (gO) are linked together, with gH and gL being involved in activating the fusogenic activity of gB, and gO working as a co-receptor. Since its discovery in the middle of the last century, hCMV has been cultivated in fibroblast cell cultures [22]. The discovery of hCMV variants that were no longer able to infect leukocytes and endothelial cells happened at the beginning of the 2000s, leading to the hypothesis that a mutation occurred in laboratory-cultivated strains. This was the starting point of the studies about genetic determinants of hCMV cell tropism [22]. A study carried out by Italian and German researchers in 2002 highlighted the role of the UL128, UL130, and UL131 locus of the hCMV genome in virus growth in endothelial cells and virus transfer to leukocytes [23]. Wang et al. [24] documented the presence of another protein complex, composed of gH/gL combined with UL128, UL130, and UL131 proteins (pUL128–pUL131), different from TC, required to infect epithelial and endothelial cells; they also found that antibodies to either protein of this pentameric structure neutralized the ability of hCMV to infect endothelial and epithelial cells but not fibroblasts. Another study showed that hCMV enters epithelial and endothelial cells by endocytosis followed by low-pH-dependent fusion, which is different from the pH-independent fusion with human fibroblasts [25]. The mechanism was definitively explained in 2008: it was shown that the transition of gH/gL-containing complexes from the endoplasmic reticulum to the Golgi apparatus and cell surface was significantly improved when all three proteins (pUL128–pUL131) were linked to gH/gL, to produce the pentameric complex (PC) gH/gL/pUL128–131 [26]. Another essential step was the discovery of the human receptors of hCMV glycoproteic complexes: platelet-derived growth factor-α receptor, as the cellular receptor of TC [27], and neutropilin2 as the PC receptor in epithelial and endothelial cells [28]. More than half of the proteins produced by hCMV are tegument phosphorylated proteins (pps). After entering the host cell, tegument pps become active, and play important roles in the various stages of the viral life cycle, such as pp65 in immune evasion, pp71 in gene expression, and pp150 and pp28 in virus assembly and egress [19][29]. The expression of herpesviruses genes proceeds with an accurate temporal order consisting of immediate-early (IE), early (E), and late (L) genes [30]. IE genes of hCMV are the first genes expressed after infection and prepare the cell for viral replication [31]. The major immediate-early (MIE) gene seems to have a role in acute infection and is an indicator of viral reactivation. The leading proteins expressed from this region are IE1 and IE2 nuclear phosphoproteins, which regulate transcription and have a role in contrasting the immune response of the host: IE1 antagonizes apoptosis and type I IFN signaling, while IE2 inhibits apoptosis and inflammatory cytokine induction [32]. The role of the various parts of the immune system in controlling hCMV infection has not yet been completely clarified but both innate and adaptive immunity seems to contribute [21].
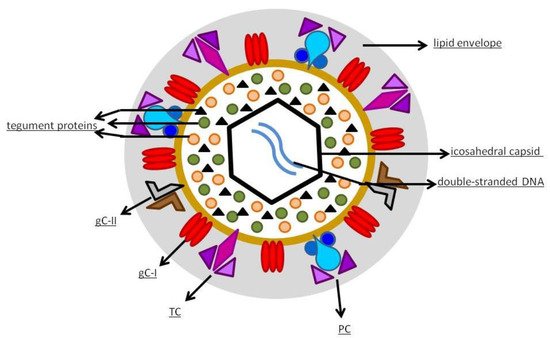
hCMV structure.
3. Clinical Manifestations
In the healthy host, hCMV infection is frequently asymptomatic; rarely, it may present as a febrile illness, or a mononucleosis-like syndrome characterized by fever, lymphadenopathy, and lymphocytosis [1]. Clinical manifestations are overt in congenital disease, transplant, and AIDS patients. In case of cCMV infection, the earliest signs of disease may be seen on fetal anatomy ultrasound at 20 weeks of gestation, even if the sensitivity is lower than 25%. In utero ultrasound indicators include echogenic bowel, brain calcifications and enlarged ventricles, neuronal migration and white matter disorders, microcephaly, fetal growth restriction, oligohydramnios or polyhydramnios, hepatosplenomegaly or hepatic calcifications, ascites, and hydrops [33].
At birth, only 10–15% of newborns with cCMV are symptomatic [34]. The clinical manifestations of cCMV at birth widely vary. Most symptomatic infants are born prematurely, <37 weeks of gestational age [35]. Common manifestations include petechiae, jaundice, hepatosplenomegaly, intrauterine growth restriction, microcephaly [36][37], hypotonia, and seizures, but also abnormal brain imaging findings, sensorineural hearing loss, and chorioretinitis can be associated [37]. Typical laboratory findings are elevated transaminases, thrombocytopenia, and an increase of direct serum bilirubin [35][37]. Permanent sequelae will develop in approximately 45% to 58% of symptomatic newborns and are predominantly motor/cognitive deficits (43%), sensorineural hearing loss (35%), and vision impairment (6%) isolated or in association [38][39][40]. On the other hand, also 10–15% of asymptomatic newborns will have long-term sequelae, mostly auditory disorders [38]. cCMV is the leading cause of non-genetic hearing impairment in children [41], and it is estimated that almost 25% of pediatric hearing loss is associated with cCMV [42]. Indeed, hypoacusia may start within the first years of life, with a median age of onset of 33 months for symptomatic and 44 months for asymptomatic infants [43]. Moreover, it can fluctuate at subsequent examinations, and it may show progressive deterioration [38][44]. The mechanism of hCMV-induced sensorineural hearing loss has not yet been well understood. Huang et al. analyzed the factors that could be responsible for this complication, including the interaction of hCMV with the Wnt and Notch signaling pathways, involved in inner ear development. They highlighted the need to further investigate this issue to provide new directions for therapeutic development [45].
In AIDS patients, the virus causes mostly sight-threatening retinitis that commonly occurs when the CD4+ T-cell count falls below 50 cells/mm3. Less frequently, it is associated with polyradiculopathy, meningoencephalitis, pneumonitis (often in co-infection with
or
), and gastrointestinal tract infections [46].
SOT recipients can develop primary or secondary hCMV infection. Manifestations can vary from myelosuppression, gastrointestinal tract, and central nervous system invasive disease, transplanted organ infection, to acute and chronic allograft injury [47]. Similarly, also in HSCT recipients, hCMV may cause a primary or a non-primary infection. Allogeneic-HSCT recipients are at higher risk than autologous-HSCT ones [48]. Clinical manifestations are similar to those of SOT patients; hCMV pneumonia is frequent in allogeneic HSCT patients, with an incidence rate of 10–30% [49].
4. Diagnosis
4.1. cCMV
Primary maternal infections can be diagnosed by serological tests, considering that the presence of hCMV-specific IgM antibodies indicate acute infection. Since IgM can persist for several months from their appearance, the clinician should test IgG avidity in case of both IgM- and IgG-positive antibodies [50]. Low-to-moderate IgG avidity is typically found for 16–18 weeks following primary infection. Consequently, positive IgM antibodies associated with low IgG avidity are suggestive of infection within the preceding 3 months, while high IgG avidity is indicative of late primary response or non-primary immune response, even if IgM antibodies are also present [51]. Maternal hCMV serology is most useful in the first trimester because of the higher risk of disease in newborns when primary infection occurs in the early stages of pregnancy [52]. However, the majority of cCMV infections are caused by non-primary maternal infection [8]. Universal prenatal screening by testing the serology of pregnant women is not routinely recommended, as there is no currently effective specific treatment to prevent transmission, it is difficult to predict sequelae and an incorrect counseling and interpretation of serology can lead to anxiety, supplementary tests, and unnecessary abortion [53][54]. In case of positive maternal serology, amniocentesis to search for hCMV DNA can be performed [55], because the urine of the infected fetus contained in the amniotic fluid could be positive to hCMV DNA. The sensitivity and specificity of this method reach their maximum when used after 17–20 weeks of gestation and 8 weeks after maternal infection [56].
Postnatal diagnosis in newborns is preferably performed via real-time polymerase chain reaction (PCR) on urine or saliva specimens [57][58]. Indeed, infected infants usually eliminate great quantities of virus in saliva and urine; thus, these samples are both suitable for the documentation of cCMV, even if false-positive tests have been reported for saliva [59] and urine collection using a bag may be difficult (e.g., inadequate diuresis, loss of sample, or contamination) [60]. Universal neonatal cCMV screening is not performed, but in the UK, Belgium, Australia, and in some states of the USA, targeted screening of infants who failed the neonatal hearing screening has been trailed [52][61][62]. However, newborns with late-onset hypoacusia would not be identified by this targeted screening. Other methods to diagnose cCMV have been studied, including PCR on dried bloodspot samples that are routinely obtained from newborns for other diagnostic purposes. This technique showed low sensitivity [60]; therefore, it is not suitable for cCMV diagnosis. However, as the dried samples can be stored for several months, it can be used for retrospective investigation of a possible vertical transmission in infants with cCMV infection, although a negative result does not exclude cCMV disease with a delayed onset of signs and symptoms [63].
4.2. Allogeneic HSCT
hCMV pp65 antigenemia assays and the hCMV DNA PCR are currently the most used laboratory techniques for the detection of hCMV infection in this kind of patient [64][65].
4.3. SOT
Performing pre-transplantation donor and recipient hCMV IgG serology is recommended, since these serostatuses are key predictors of the risk of hCMV infection after transplant and guide decisions on antiviral prophylaxis or pre-emptive therapy [66]. Serology has no role in the diagnosis of active hCMV replication and disease post-transplantation. Quantitative nucleic acid amplification testing is the preferred method for diagnosis of hCMV infection [66].
5. Therapy
5.1. cCMV
At present, antiviral therapy is only indicated for cCMV symptomatic patients, since there is no evidence of benefit of treatment in asymptomatic neonates. In symptomatic cCMV, antiviral drugs (intravenous (IV) Ganciclovir 6 mg/kg two times a day for 6 weeks or oral Valganciclovir 16 mg/kg two times a day for 6 months) are effective on hearing and neurodevelopmental long-term outcomes [67][68][69][70]. In a retrospective study, Bilavsky et al. observed that infants born with cCMV and hearing impairment receiving Ganciclovir or Valganciclovir for 12 months showed significant improvement in their hearing status [40]. Adverse effects are common in neonates treated for cCMV infection. The main side effect of antiviral therapy is neutropenia, which occurs in approximately 50% of infants and is more common with IV Ganciclovir than with oral Valganciclovir [69][71]. It generally occurs in the first month of treatment, so no increased toxicity was observed in randomized control trials evaluating 6 months vs. 6 weeks of treatment. Neutropenia is rarely severe and usually resolves with dose adjustment, administration of granulocyte colony-stimulating factor (G-CSF), or treatment discontinuation [71][72]. Other common side effects are thrombocytopenia and hepatotoxicity, reported in up to 30% of patients treated with Ganciclovir. Considering antiviral toxicity, it is important to monitor patients regularly with clinical examination and blood tests.
5.2. Allogeneic HSCT
Pre-emptive treatment refers to immediate antiviral administration when hCMV antigenemia or viremia first occurs following transplantation [64]. Ganciclovir is used as pre-emptive therapy in transplant patients, as well as Valganciclovir, Foscarnet, and Cidofovir [64]. Pre-emptive treatment should be maintained until the relevant symptoms are resolved and the hCMV serum test is negative. Subsequently, patients should receive maintenance treatment for a variable period [73]. The length of maintenance treatment varies from 0–6 weeks depending on many factors (such as patients’ sensitivity to treatment, drug side effects, and risk of relapse [74]).
In HSCT symptomatic patients, Ganciclovir is the drug of choice for early treatment but may have a myelosuppressive effect, which can be improved by G-CSF alone or in combination with anti-CMV immunoglobulins. Guidelines recommend induction with IV Gangiclovir 5 mg/kg/die administered twice a day for 7–14 days, followed by maintenance therapy once a day until two consecutive tests are negative [75].
Foscarnet demonstrated similar effects to Ganciclovir but did not provoke granulocytopenia, making it suitable for patients with bone marrow suppression [76]. The main adverse reaction is electrolyte imbalance. An additional drug used in pre-emptive treatment is Cidofovir; its administration is weekly, and its main side effect is renal toxicity. Cidofovir is often administered when other treatments have previously been ineffective or in case of intolerance to Ganciclovir or Foscarnet [77]. Letermovir is a novel antiviral drug that suppresses the hCMV-terminase complex [78].
5.3. SOT
Prophylaxis causes a delayed onset of hCMV disease and reduced complications [79]. Valganciclovir is currently used for this purpose, starting within 10 days after transplant and continuing for 3–12 months [66][80]. In asymptomatic patients, once viremia reaches the positivity threshold, it is recommended to start treatment with Valganciclovir (900 mg every 12 h) and to continue until viremia is negative, with a minimum of 2 weeks of treatment [66]. In case of symptomatic hCMV infection, treatment with Valganciclovir 900 mg every 12 h or IV Ganciclovir 5 mg/kg every 12 h is recommended. This treatment should be continued for a minimum of 2 weeks, until clinical resolution and eradication of hCMV [66].
5.4. AIDS
In AIDS patients, antiviral medications against hCMV are used to treat hCMV retinitis: they include systemic (IV Ganciclovir, oral Valganciclovir, IV Foscarnet, or IV Cidofovir) and intravitreal therapy (Ganciclovir, Foscarnet, or Cidofovir) [81].
6. Why Is It Necessary to Find an Effective Vaccine against hCMV?
hCMV-related disease has a significant impact both on patients’ lives and on health care costs. In transplant populations, morbidity from hCMV is extremely relevant, and antiviral prophylaxis is expensive, not completely successful, and has a limited duration [82]. Every year, thousands of children suffer the consequences of cCMV disease: they are afflicted by permanent disabilities, such as hearing loss, vision loss, and motor and cognitive deficits [83]. Retzler et al. tried to estimate the annual economic burden of managing cCMV and its sequelae in the UK [84]. Their model calculated that the total cost of cCMV to the UK in 2016 was 732 million pounds, 40% of the costs being charged to the public sector while the remaining 60% being indirect costs, such as lost productivity. Moreover, long-term sequelae were reported having a higher financial burden than the acute management. All things considered, a vaccine against hCMV would be highly cost effective [85]. Moreover, hCMV can manipulate the immune system and this seems to compromise self-tolerance in genetically predisposed individuals. It appears that hCMV can lead to immune dysregulation, which triggers the initiation or perpetuation of autoimmune diseases, such as systemic lupus erythematosus and rheumatoid arthritis, with mechanisms, such as molecular mimicry, inflammation, and nonspecific B cell activation [86]. Because of the great commitment required of the immune system to keep the virus silent, there is also growing evidence that hCMV may be the underlying cause of immune senescence and cardiovascular diseases, including atherosclerosis, ischemic heart disease, myocardial infarction, and cardiovascular death [87][88]. Another open issue is the possibility that the presence of hCMV in the host contributes to tumor growth or progression. Recent evidence suggests that hCMV might play a causative role in the pathogenesis and progression of glioblastoma multiforme, colon cancer, and infant leukemia [89].
There is therefore a need to clarify all other possible manifestations and implications of hCMV infection, because this could lead to consideration of universal immunization against hCMV, although this seems a difficult road [90]. Researchers’ and pharmaceutical companies’ attention was brought to this subject in 2000, after the publication of a vaccine priority document written by the National Academy of Medicine of United States that put hCMV vaccine in the group with the highest priority for development [91].
7. Where Are We Now?
Despite the clear need, progress toward a vaccine for hCMV has been slow. The first attempts to create a hCMV vaccine began in the 1970s, when two strains of the virus, Towne and AD169, were attenuated in order to function as active immunoprophylaxis [92][93]. The project with the Towne strain was developed by Stanley Plotkin and provided clinical studies with SOT recipients: initial results seemed promising, but statistical analyses revealed that protection against infection was not significant [94][95]. Subsequently, a growing number of hCMV vaccine candidates were developed and are now at different stages, but to date, none have been licensed. There are many intrinsic features of hCMV that are challenging for the design of a vaccine. It can cause persistent asymptomatic infection and establish a lifelong latency in the host after subclinical primary infection [85]; it is able to spread cell–cell, avoiding antibodies in the extracellular fluid [96]; moreover, hCMV reactivations can happen during periods of decreased immune system defenses [85]. Another critical consideration concerns interstrain diversity: hCMV undergoes pervasive recombination with disruptive mutations identified in clinical isolates [97], even with rapid intra-host evolution [98], so that reinfections can occur with different strains [99]. Moreover, hCMV proliferation in the host is species restricted so there are no natural models available to test vaccine strategies [85].
It is well established that children born with congenital infection and immunocompromised subjects are the two groups of patients suffering the most serious consequences after contact with hCMV. Thus, despite the ideal target populations still being controversial at present, it seems reasonable that the more suitable could be pregnant women or women of childbearing age and subjects undergoing transplant [100]. Regarding the vaccination of transplant recipients, the major challenge is inducing an adequate immunity in an immunocompromised individual. It seems that both T cells and neutralizing antibodies are implicated and should be stimulated by a hCMV vaccine in this target population [101]. Concerning cCMV, considering the epidemiological characteristics and modality of vertical transmission, the vaccine should be able to protect both seronegative women from primary infection and seropositive women from reinfection and reactivation [100]. It has been also proposed to include the hCMV vaccine into the routine childhood vaccination schedule, supporting universal vaccination, bearing in mind that the hCMV infection implications are not yet completely clear and that many women are infected by their own children or during jobs that require close contact with children, so the vaccination of toddlers would probably provide indirect protection [82]. With regards to cCMV, we still do not know the exact details of immune mechanisms against this virus, but most data indicate that the vaccine should stimulate both the cellular and humoral components [102]. After a primary maternal hCMV infection in the first trimester, hyperimmunoglobulin administration apparently prevents maternal–fetal transmission, implicitly suggesting a role of antibodies [103], and CD4+ T cells have been correlated with protection against hCMV [104][105]. Considering that infants born to mothers who underwent reinfection or reactivation during pregnancy are still at risk for congenital disease, and that we do not know the specific contributions of humoral and cellular immunity for the prevention of this condition, it could seem very difficult to create an effective vaccine for avoiding vertical transmission [90]. A recent systematic review [5], however, confirmed that the hCMV placental transmission rate is lower in non-primary than in primary infection, with low cCMV infection prevalence in highly seropositive populations. In addition, Tabata et al. [106] showed that human monoclonal antibodies to gB and PC prevent infection in placental cells and anchoring villi better than hyperimmunoglobulin. This evidence might prove that maternal immunity confers protection and pregnant women and women of child-bearing age could be a good target for a hCMV vaccine. Efforts to produce the vaccine have focused on few antigens; neutralizing antibody targets, such as gB, gH, and PC; and T cell epitopes, such as pp65 and IE1 [102]. Initially, gB seemed a perfect choice, but trials showed limited efficacy [102]. At present, not only neutralization but also other antibody mechanisms are known to play a role, such as the induction of complement-mediated virus lysis and antibody-dependent cellular cytotoxicity [102]. A very important step for the development of hCMV vaccine is represented by the identification of new viral glycoproteins and cellular receptors implicated in virus entry in host cells, as previously described [22]. This new awareness led to investigation of the humoral response to hCMV infection, to understand which of the viral antigens could be more important to obtain protective antibodies and to be used in a vaccine [22]. Fouts et al. studied the anti-CMV hyperimmune globulin used to prevent hCMV disease in SOT patients and cCMV and found that the most neutralizing response was provided by antibodies directed against PC, with a little role of anti-gB antibodies [107]. Similar conclusions were also drawn from other studies [108][109], thus supporting the development of PC-based vaccines. To date, the most promising choice in developing hCMV vaccines seems to be an approach that requires the expression of several antigens, with epitopes able to stimulate both the humoral and the cellular components; moreover, the role of protein conformation and structural biology in elicitation of the right immune response has been recognized, as in the case of the pre- and post-fusion crystal structure of gB [102]. Consequently, mRNA and vector vaccines, able to simultaneously synthesize a combination of antigens in their original three-dimensional conformation, could represent good candidates [102]. Chauhan and Singh recently described an immuno-informatics approach to design a multi-epitope vaccine [110]. In the study, a multi-epitope vaccine was constructed thanks to immuno-informatics servers, which selected the most suitable epitopes of PC, gB, and pp65. The results showed that the vaccine might be immunogenic, with high affinity with the immune receptor, effective in stimulating different immune cell types, and could provide long-lasting immune response.
References
- Bate, S.L.; Dollard, S.C.; Cannon, M.J. Cytomegalovirus Seroprevalence in the United States: The National Health and Nutrition Examination Surveys, 1988. Clin. Infect. Dis. 2010, 50, 1439–1447.
- Gerna, G.; Lilleri, D. Human Cytomegalovirus (HCMV) Infection/Re-Infection: Development of a Protective HCMV Vaccine. New Microbiol. 2019, 42, 1–20.
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276.
- Cannon, M.J.; Hyde, T.B.; Schmid, D.S. Review of cytomegalovirus shedding in bodily fluids and relevance to congenital cytomegalovirus infection. Rev. Med. Virol. 2011, 21, 240–255.
- Coppola, T.; Mangold, J.F.; Cantrell, S.; Permar, S.R. Impact of Maternal Immunity on Congenital Cytomegalovirus Birth Prevalence and Infant Outcomes: A Systematic Review. Vaccines 2019, 7, 129.
- Revello, M.G.; Fabbri, E.; Furione, M.; Zavattoni, M.; Lilleri, D.; Tassis, B.; Quarenghi, A.; Cena, C.; Arossa, A.; Montanari, L.; et al. Role of prenatal diagnosis and counseling in the management of 735 pregnancies complicated by primary human cytomegalovirus infection: A 20-year experience. J. Clin. Virol. 2011, 50, 303–307.
- Boppana, S.B.; Rivera, L.B.; Fowler, K.B.; Mach, M.; Britt, W.J. Intrauterine Transmission of Cytomegalovirus to Infants of Women with Preconceptional Immunity. N. Engl. J. Med. 2001, 344, 1366–1371.
- De Vries, J.J.C.; Van Zwet, E.W.; Dekker, F.W.; Kroes, A.C.M.; Verkerk, P.H.; Vossen, A.C.T.M. The apparent paradox of maternal seropositivity as a risk factor for congenital cytomegalovirus infection: A population-based prediction model. Rev. Med. Virol. 2013, 23, 241–249.
- Enders, G.; Daiminger, A.; Bäder, U.; Exler, S.; Enders, M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J. Clin. Virol. 2011, 52, 244–246.
- Pass, R.F.; Fowler, K.B.; Boppana, S.B.; Britt, W.J.; Stagno, S. Congenital cytomegalovirus infection following first trimester maternal infection: Symptoms at birth and outcome. J. Clin. Virol. 2006, 35, 216–220.
- Fishman, J.A. Infection in Solid-Organ Transplant Recipients. N. Engl. J. Med. 2007, 357, 2601–2614.
- Hoover, D.R.; Saah, A.J.; Bacellar, H.; Phair, J.; Detels, R.; Anderson, R.; Kaslow, R.A. Clinical Manifestations of AIDS in the Era of Pneumocystis Prophylaxis. N. Engl. J. Med. 1993, 329, 1922–1926.
- Sungkanuparph, S.; Chakriyanuyok, T.; Butthum, B. Antiretroviral therapy in AIDS patients with CMV disease: Impact on the survival and long-term treatment outcome. J. Infect. 2008, 56, 40–43.
- Cervera, C.; Gurgui, M.; Lumbreras, C. Factores de riesgo de la enfermedad por citomegalovirus en el receptor de un trasplante de órgano sólido. Enfermedades Infec. Microbiol. Clín. 2011, 29, 11–17.
- Bermejo, C.L.; Manuel, O.; Len, O.; ten Berge, I.J.T.; Sgarabotto, D.; Hirsch, H.H. Cytomegalovirus infection in solid organ transplant recipients. Clin. Microbiol. Infect. 2014, 20 (Suppl. 7), 19–26.
- Chan, S.T.; Logan, A.C. The clinical impact of cytomegalovirus infection following allogeneic hematopoietic cell transplantation: Why the quest for meaningful prophylaxis still matters. Blood Rev. 2017, 31, 173–183.
- Owers, D.S.; Webster, A.C.; Strippoli, G.F.M.; Kable, K.; Hodson, E.M. Pre-emptive treatment for cytomegalovirus viraemia to prevent cytomegalovirus disease in solid organ transplant recipients. Cochrane Database Syst. Rev. 2013, CD005133.
- Limaye, A.P.; Boeckh, M. CMV in critically ill patients: Pathogen or bystander? Rev. Med. Virol. 2010, 20, 372–379.
- Iii, J.P.T. Human cytomegalovirus tegument proteins (pp65, pp71, pp150, pp28). Virol. J. 2012, 9, 22.
- Human Cytomegalovirus; Shenk, T.; Stinski, M. Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2008.
- Sandonís, V.; García-Ríos, E.; McConnell, M.J.; Pérez-Romero, P. Role of Neutralizing Antibodies in CMV Infection: Implications for New Therapeutic Approaches. Trends Microbiol. 2020, 28, 900–912.
- Gerna, G.; Kabanova, A.; Lilleri, D. Human Cytomegalovirus Cell Tropism and Host Cell Receptors. Vaccines 2019, 7, 70.
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human Cytomegalovirus UL131-128 Genes Are Indispensable for Virus Growth in Endothelial Cells and Virus Transfer to Leukocytes. J. Virol. 2004, 78, 10023–10033.
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158.
- Ryckman, B.J.; Jarvis, M.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human Cytomegalovirus Entry into Epithelial and Endothelial Cells Depends on Genes UL128 to UL150 and Occurs by Endocytosis and Low-pH Fusion. J. Virol. 2006, 80, 710–722.
- Ryckman, B.J.; Rainish, B.L.; Chase, M.C.; Borton, J.A.; Nelson, J.A.; Jarvis, M.; Johnson, D.C. Characterization of the Human Cytomegalovirus gH/gL/UL128-131 Complex That Mediates Entry into Epithelial and Endothelial Cells. J. Virol. 2007, 82, 60–70.
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M.; Fernandez-Rodriguez, B.M.; et al. Platelet-derived growth factor-α receptor is the cellular receptor for human cytomegalovirus gHgLgO trimer. Nat. Microbiol. 2016, 1, 16082.
- Martinez-Martin, N.; Marcandalli, J.; Huang, C.S.; Arthur, C.P.; Perotti, M.; Foglierini, M.; Ho, H.; Dosey, A.M.; Shriver, S.; Payandeh, J.; et al. An Unbiased Screen for Human Cytomegalovirus Identifies Neuropilin-2 as a Central Viral Receptor. Cell 2018, 174, 1158–1171.e19.
- Kalejta, R.F. Tegument Proteins of Human Cytomegalovirus. Microbiol. Mol. Biol. Rev. 2008, 72, 249–265.
- Gugliesi, F.; Coscia, A.; Griffante, G.; Galitska, G.; Pasquero, S.; Albano, C.; Biolatti, M. Where do we Stand after Decades of Studying Human Cytomegalovirus? Microorganisms 2020, 8, 685.
- Stenberg, R.M. Immediate-Early Genes of Human Cytomegalovirus: Organization and Function. In Molecular Aspects of Human Cytomegalovirus Diseases; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 1993; Volume 2, pp. 330–359.
- Paulus, C.; Nevels, M. The Human Cytomegalovirus Major Immediate-Early Proteins as Antagonists of Intrinsic and Innate Antiviral Host Responses. Viruses 2009, 1, 760–779.
- Guerra, B.; Simonazzi, G.; Puccetti, C.; Lanari, M.; Farina, A.; Lazzarotto, T.; Rizzo, N. Ultrasound prediction of symptomatic congenital cytomegalovirus infection. Am. J. Obstet. Gynecol. 2008, 198, 380.e1–380.e7.
- Thigpen, J. Congenital Cytomegalovirus—History, Current Practice, and Future Opportunities. Neonatal Netw. 2020, 39, 293–298.
- Boppana, S.B.; Ross, S.A.; Fowler, K.B. Congenital Cytomegalovirus Infection: Clinical Outcome. Clin. Infect. Dis. 2013, 57, S178–S181.
- Kylat, R.I.; Kelly, E.N.; Ford-Jones, E.L. Clinical findings and adverse outcome in neonates with symptomatic congenital cytomegalovirus (SCCMV) infection. Eur. J. Nucl. Med. Mol. Imaging 2006, 165, 773–778.
- Dreher, A.M.; Arora, N.; Fowler, K.B.; Novak, Z.; Britt, W.J.; Boppana, S.B.; Ross, S.A. Spectrum of Disease and Outcome in Children with Symptomatic Congenital Cytomegalovirus Infection. J. Pediatr. 2014, 164, 855–859.
- Goderis, J.; De Leenheer, E.; Smets, K.; Van Hoecke, H.; Keymeulen, A.; Dhooge, I. Hearing Loss and Congenital CMV Infection: A Systematic Review. Pediatry 2014, 134, 972–982.
- Cannon, M.J.; Griffiths, P.D.; Aston, V.; Rawlinson, W.D. Universal newborn screening for congenital CMV infection: What is the evidence of potential benefit? Rev. Med. Virol. 2014, 24, 291–307.
- Bilavsky, E.; Shahar-Nissan, K.; Pardo, J.; Attias, J.; Amir, J. Hearing outcome of infants with congenital cytomegalovirus and hearing impairment. Arch. Dis. Child. 2016, 101, 433–438.
- Grosse, S.D.; Ross, D.S.; Dollard, S.C. Congenital cytomegalovirus (CMV) infection as a cause of permanent bilateral hearing loss: A quantitative assessment. J. Clin. Virol. 2008, 41, 57–62.
- Morton, C.C.; Nance, W.E. Newborn Hearing Screening—A Silent Revolution. N. Engl. J. Med. 2006, 354, 2151–2164.
- Dahle, A.J.; Fowler, K.B.; Wright, J.D.; Boppana, S.B.; Britt, W.J.; Pass, R. Longitudinal investigation of hearing disorders in children with congenital cytomegalovirus. J. Am. Acad. Audiol. 2000, 11, 283–290.
- Foulon, I.; Naessens, A.; Foulon, W.; Casteels, A.; Gordts, F. A 10-Year Prospective Study of Sensorineural Hearing Loss in Children with Congenital Cytomegalovirus Infection. J. Pediatr. 2008, 153, 84–88.
- Huang, S.-N.; Zhou, Y.-P.; Jiang, X.; Yang, B.; Cheng, H.; Luo, M.-H. Hearing Loss Caused by HCMV Infection through Regulating the Wnt and Notch Signaling Pathways. Viruses 2021, 13, 623.
- Dioverti, M.V.; Razonable, R.R. Cytomegalovirus. Microbiol. Spectr. 2016, 4, 97–125.
- Razonable, R.R.; Humar, A.; the AST Infectious Diseases Community of Practice. Cytomegalovirus in Solid Organ Transplantation. Arab. Archaeol. Epigr. 2013, 13, 93–106.
- George, B.; Pati, N.; Gilroy, N.; Ratnamohan, M.; Huang, G.; Kerridge, I.; Hertzberg, M.; Gottlieb, D.; Bradstock, K. Pre-transplant cytomegalovirus (CMV) serostatus remains the most important determinant of CMV reactivation after allogeneic hematopoietic stem cell transplantation in the era of surveillance and preemptive therapy. Transpl. Infect. Dis. 2010, 12, 322–329.
- Travi, G.; Pergam, S.A. Cytomegalovirus Pneumonia in Hematopoietic Stem Cell Recipients. J. Intensive Care Med. 2014, 29, 200–212.
- Prince, H.E.; Lapé-Nixon, M. Role of Cytomegalovirus (CMV) IgG Avidity Testing in Diagnosing Primary CMV Infection during Pregnancy. Clin. Vaccine Immunol. 2014, 21, 1377–1384.
- Lazzarotto, T.; Varani, S.; Spezzacatena, P.; Gabrielli, L.; Pradelli, P.; Guerra, B.; Landini, M.P. Maternal IgG Avidity and IgM Detected by Blot as Diagnostic Tools to Identify Pregnant Women at Risk of Transmitting Cytomegalovirus. Viral Immunol. 2000, 13, 137–141.
- Lazzarotto, T.; Blázquez-Gamero, D.; Delforge, M.-L.; Foulon, I.; Luck, S.; Modrow, S.; Leruez-Ville, M. Congenital Cytomegalovirus Infection: A Narrative Review of the Issues in Screening and Management from a Panel of European Experts. Front. Pediatr. 2020, 8, 13.
- Rawlinson, W.D.; Boppana, S.B.; Fowler, K.B.; Kimberlin, D.W.; Lazzarotto, T.; Alain, S.; Daly, K.; Doutré, S.; Gibson, L.; Giles, M.L.; et al. Congenital cytomegalovirus infection in pregnancy and the neonate: Consensus recommendations for prevention, diagnosis, and therapy. Lancet Infect. Dis. 2017, 17, e177–e188.
- Luck, S.E.; Wieringa, J.W.; Blázquez-Gamero, D.; Henneke, P.; Schuster, K.; Butler, K.; Capretti, M.G.; Cilleruelo, M.J.; Curtis, N.; Garofoli, F.; et al. Congenital Cytomegalovirus. Pediatr. Infect. Dis. J. 2017, 36, 1205–1213.
- Hughes, B.L.; Gyamfi-Bannerman, C. Diagnosis and antenatal management of congenital cytomegalovirus infection. Am. J. Obstet. Gynecol. 2016, 214, B5–B11.
- Enders, M.; Daiminger, A.; Exler, S.; Ertan, K.; Enders, G.; Bald, R. Prenatal diagnosis of congenital cytomegalovirus infection in 115 cases: A 5 years’ single center experience. Prenat. Diagn. 2017, 37, 389–398.
- Boppana, S.B.; Ross, S.A.; Shimamura, M.; Palmer, A.L.; Ahmed, A.; Michaels, M.G.; Sánchez, P.J.; Bernstein, D.I.; Tolan, R.W.; Novak, Z.; et al. Saliva Polymerase-Chain-Reaction Assay for Cytomegalovirus Screening in Newborns. N. Engl. J. Med. 2011, 364, 2111–2118.
- de Vries, J.J.; van der Eijk, A.A.; Wolthers, K.C.; Rusman, L.G.; Pas, S.D.; Molenkamp, R.; Claas, E.C.; Kroes, A.C.; Vossen, A.C. Real-time PCR vs. viral culture on urine as a gold standard in the diagnosis of congenital cytomegalovirus infection. J. Clin. Virol. 2012, 53, 167–170.
- Leruez-Ville, M.; Magny, J.-F.; Couderc, S.; Pichon, C.; Parodi, M.; Bussières, L.; Guilleminot, T.; Ghout, I.; Ville, Y. Risk Factors for Congenital Cytomegalovirus Infection Following Primary and Nonprimary Maternal Infection. Clin. Infect. Dis. 2017, 65, 398–404.
- Ross, S.A.; Ahmed, A.; Palmer, A.L.; Michaels, M.G.; Sánchez, P.J.; Stewart, A.; Bernstein, D.I.; Feja, K.; Novak, Z.; Fowler, K.B.; et al. Urine Collection Method for the Diagnosis of Congenital Cytomegalovirus Infection. Pediatr. Infect. Dis. J. 2015, 34, 903–905.
- Vancor, E.; Shapiro, E.D.; Loyal, J. Results of a Targeted Screening Program for Congenital Cytomegalovirus Infection in Infants Who Fail Newborn Hearing Screening. J. Pediatr. Infect. Dis. Soc. 2018, 8, 55–59.
- Diener, M.L.; Zick, C.D.; McVicar, S.B.; Boettger, J.; Park, A.H. Outcomes from a Hearing-Targeted Cytomegalovirus Screening Program. Pediatry 2017, 139, e20160789.
- Marsico, C.; Kimberlin, D.W. Congenital Cytomegalovirus infection: Advances and challenges in diagnosis, prevention and treatment. Ital. J. Pediatr. 2017, 43, 1–8.
- Drew, W.L. Laboratory diagnosis of cytomegalovirus infection and disease in immunocompromised patients. Curr. Opin. Infect. Dis. 2007, 20, 408–411.
- Zhou, X.; Jin, N.; Chen, B. Human cytomegalovirus infection: A considerable issue following allogeneic hematopoietic stem cell transplantation (Review). Oncol. Lett. 2021, 21, 1–15.
- Kotton, C.N.; Kumar, D.; Caliendo, A.M.; Huprikar, S.; Chou, S.; Danziger-Isakov, L.; Humar, A. The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-organ Transplantation. Transplant 2018, 102, 900–931.
- Kimberlin, D.W.; Acosta, E.P.; Sánchez, P.J.; Sood, S.; Agrawal, V.; Homans, J.; Jacobs, R.F.; Lang, D.; Romero, J.R.; Griffin, J.; et al. Pharmacokinetic and Pharmacodynamic Assessment of Oral Valganciclovir in the Treatment of Symptomatic Congenital Cytomegalovirus Disease. J. Infect. Dis. 2008, 197, 836–845.
- Kimberlin, D.W.; Lin, C.-Y.; Sánchez, P.J.; Demmler, G.J.; Dankner, W.; Shelton, M.; Jacobs, R.F.; Vaudry, W.; Pass, R.; Kiell, J.M.; et al. Effect of ganciclovir therapy on hearing in symptomatic congenital cytomegalovirus disease involving the central nervous system: A randomized, controlled trial. J. Pediatr. 2003, 143, 16–25.
- Kimberlin, D.W.; Jester, P.M.; Sánchez, P.J.; Ahmed, A.; Arav-Boger, R.; Michaels, M.G.; Ashouri, N.; Englund, J.A.; Estrada, B.; Jacobs, R.F.; et al. Valganciclovir for Symptomatic Congenital Cytomegalovirus Disease. N. Engl. J. Med. 2015, 372, 933–943.
- Oliver, S.E.; Cloud, G.A.; Sánchez, P.J.; Demmler, G.J.; Dankner, W.; Shelton, M.; Jacobs, R.F.; Vaudry, W.; Pass, R.; Soong, S.-J.; et al. Neurodevelopmental outcomes following ganciclovir therapy in symptomatic congenital cytomegalovirus infections involving the central nervous system. J. Clin. Virol. 2009, 46, S22–S26.
- Gwee, A.; Curtis, N.; Connell, T.G.; Garland, S.; Daley, A.J. Ganciclovir for the Treatment of Congenital Cytomegalovirus. Pediatr. Infect. Dis. J. 2014, 33, 115.
- Lombardi, G.; Garofoli, F.; Villani, P.; Tizzoni, M.; Angelini, M.; Cusato, M.; Bollani, L.; De Silvestri, A.; Regazzi, M.; Stronati, M. Oral valganciclovir treatment in newborns with symptomatic congenital cytomegalovirus infection. Eur. J. Clin. Microbiol. Infect. Dis. 2009, 28, 1465–1470.
- Park, S.-Y.; Lee, S.-O.; Choi, S.-H.; Kim, Y.S.; Woo, J.H.; Baek, S.; Sung, H.; Kim, M.-N.; Kim, D.-Y.; Lee, J.-H.; et al. Efficacy and safety of low-dose ganciclovir preemptive therapy in allogeneic haematopoietic stem cell transplant recipients compared with conventional-dose ganciclovir: A prospective observational study. J. Antimicrob. Chemother. 2012, 67, 1486–1492.
- Locatelli, F.; Bertaina, A.; Bertaina, V.; Merli, P. Cytomegalovirus in hematopoietic stem cell transplant recipients—management of infection. Expert Rev. Hematol. 2016, 9, 1093–1105.
- Maffini, E.; Giaccone, L.; Festuccia, M.; Brunello, L.; Busca, A.; Bruno, B. Treatment of CMV Infection after Allogeneic Hematopoietic Stem Cell Transplantation. Expert Rev. Hematol. 2016, 9, 585–596.
- Moretti, S.; Zikos, P.; Van Lint, M.T.; Tedone, E.; Occhini, D.; Gualandi, F.; Lamparelli, T.; Mordini, N.; Berisso, G.; Bregante, S.; et al. Foscarnet vs ganciclovir for cytomegalovirus (CMV) antigenemia after allogeneic hemopoietic stem cell transplantation (HSCT): A randomised study. Bone Marrow Transplant. 1998, 22, 175–180.
- Meesing, A.; Razonable, R.R. New Developments in the Management of Cytomegalovirus Infection after Transplantation. Drugs 2018, 78, 1085–1103.
- Marty, F.M.; Ljungman, P.; Chemaly, R.F.; Maertens, J.; Dadwal, S.S.; Duarte, R.F.; Haider, S.; Ullmann, A.J.; Katayama, Y.; Brown, J.; et al. Letermovir Prophylaxis for Cytomegalovirus in Hematopoietic-Cell Transplantation. N. Engl. J. Med. 2017, 377, 2433–2444.
- Paya, C.; Humar, A.; Dominguez, E.; Washburn, K.; Blumberg, E.; Alexander, B.; Freeman, R.; Heaton, N.; Pescovitz, M.D.; Valganciclovir Solid Organ Transplant Study Group. Efficacy and Safety of Valganciclovir vs. Oral Ganciclovir for Prevention of Cytomegalovirus Disease in Solid Organ Transplant Recipients. Arab. Archaeol. Epigr. 2004, 4, 611–620.
- Razonable, R.R.; Humar, A. Cytomegalovirus in Solid Organ Transplant Recipients—Guidelines of the American Society of Transplantation Infectious Diseases Community of Practice. Clin. Transplant. 2019, 33, e13512.
- Port, A.D.; Orlin, A.; Kiss, S.; Patel, S.; D’Amico, D.J.; Gupta, M.P. Cytomegalovirus Retinitis: A Review. J. Ocul. Pharmacol. Ther. 2017, 33, 224–234.
- Plotkin, S.A.; Boppana, S.B. Vaccination against the human cytomegalovirus. Vaccine 2019, 37, 7437–7442.
- Fowler, K.B.; Boppana, S.B. Congenital cytomegalovirus infection. Semin. Perinatol. 2018, 42, 149–154.
- Retzler, J.; Hex, N.; Bartlett, C.; Webb, A.; Wood, S.; Star, C.; Griffiths, P.; Jones, C.E. Economic cost of congenital CMV in the UK. Arch. Dis. Child. 2018, 104, 559–563.
- La Rosa, C.; Diamond, D.J. The immune response to human CMV. Futur. Virol. 2012, 7, 279–293.
- Gugliesi, F.; Pasquero, S.; Griffante, G.; Scutera, S.; Albano, C.; Pacheco, S.; Riva, G.; Dell’Oste, V.; Biolatti, M. Human Cytomegalovirus and Autoimmune Diseases: Where Are We? Viruses 2021, 13, 260.
- Pawelec, G.; Akbar, A.; Beverley, P.; Caruso, C.; Derhovanessian, E.; Fülöp, T.; Griffiths, P.; Grubeck-Loebenstein, B.; Hamprecht, K.; Jahn, G.; et al. Immunosenescence and Cytomegalovirus: Where do we stand after a decade? Immun. Ageing 2010, 7, 13.
- Vasilieva, E.; Gianella, S.; Freeman, M.L. Novel Strategies to Combat CMV-Related Cardiovascular Disease. Pathog. Immun. 2020, 5, 240–274.
- Wilski, N.A.; Snyder, C.M. From Vaccine Vector to Oncomodulation: Understanding the Complex Interplay between CMV and Cancer. Vaccines 2019, 7, 62.
- Schleiss, M.R.; Diamond, D.J. Exciting Times for Cytomegalovirus (CMV) Vaccine Development: Navigating the Pathways toward the Goal of Protecting Infants against Congenital CMV Infection. Vaccines 2020, 8, 526.
- Institute of Medicine (US) Committee to Study Priorities for Vaccine Development. Vaccines for the 21st Century: A Tool for Decisionmaking; Stratton, K.R., Durch, J.S., Lawrence, R.S., Eds.; The National Academies Collection: Reports Funded by National Institutes of Health; National Academies Press (US): Washington, DC, USA, 2000.
- Plotkin, S.A.; Furukawa, T.; Zygraich, N.; Huygelen, C. Candidate cytomegalovirus strain for human vaccination. Infect. Immun. 1975, 12, 521–527.
- Elek, S.; Stern, H. Development of a vaccine against mental retardation caused by cytomegalovirus infection in utero. Lancet 1974, 303, 1–5.
- Plotkin, S.A.; Starr, S.E.; Friedman, H.M.; Brayman, K.; Harris, S.; Jackson, S.; Tustin, N.B.; Grossman, R.; Dafoe, D.; Barker, C. Effect of Towne Live Virus Vaccine on Cytomegalovirus Disease after Renal Transplant. Ann. Intern. Med. 1991, 114, 525–531.
- Plotkin, S.A.; Higgins, R.; Kurtz, J.B.; Morris, P.J.; Campbell, D.A.; Shope, T.C.; Spector, S.A.; Dankner, W.M. Multi-center Trial of Towne Strain Attenuated Virus Vaccine in Seronegative Renal Transplant Recipients. Transplantation 1994, 58, 1176–1178.
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297.
- Sijmons, S.; Thys, K.; Ngwese, M.M.; Van Damme, E.; Dvorak, J.; Van Loock, M.; Li, G.; Tachezy, R.; Busson, L.; Aerssens, J.; et al. High-Throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-Disrupting Mutations and Pervasive Recombination. J. Virol. 2015, 89, 7673–7695.
- Renzette, N.; Gibson, L.; Bhattacharjee, B.; Fisher, D.; Schleiss, M.R.; Jensen, J.D.; Kowalik, T.F. Rapid Intrahost Evolution of Human Cytomegalovirus Is Shaped by Demography and Positive Selection. PLoS Genet. 2013, 9, e1003735.
- Grundy, J.; Super, M.; Sweny, P.; Moorhead, J.; Lui, S.; Berry, N.; Fernando, O.; Griffiths, P. Symptomatic cytomegalovirus infection in seropositive kidney recipients: Reinfection with donor virus rather than reactivation of recipient virus. Lancet 1988, 332, 132–135.
- Anderholm, K.M.; Bierle, C.J.; Schleiss, M.R. Cytomegalovirus Vaccines: Current Status and Future Prospects. Drugs 2016, 76, 1625–1645.
- Diamond, D.J.; La Rosa, C.; Chiuppesi, F.; Contreras, H.; Dadwal, S.; Wussow, F.; Bautista, S.; Nakamura, R.; Zaia, J.A. A fifty-year odyssey: Prospects for a cytomegalovirus vaccine in transplant and congenital infection. Expert Rev. Vaccines 2018, 17, 889–911.
- Nelson, C.S.; Herold, B.C.; Permar, S.R. A new era in cytomegalovirus vaccinology: Considerations for rational design of next-generation vaccines to prevent congenital cytomegalovirus infection. Vaccines 2018, 3, 1–9.
- Kagan, K.O.; Enders, M.; Schampera, M.S.; Baeumel, E.; Hoopmann, M.; Geipel, A.; Berg, C.; Goelz, R.; De Catte, L.; Wallwiener, D.; et al. Prevention of maternal-fetal transmission of cytomegalovirus after primary maternal infection in the first trimester by biweekly hyperimmunoglobulin administration. Ultrasound Obstet. Gynecol. 2019, 53, 383–389.
- Lilleri, D.; Fornara, C.; Furione, M.; Zavattoni, M.; Revello, M.G.; Gerna, G. Development of Human Cytomegalovirus–Specific T Cell Immunity during Primary Infection of Pregnant Women and Its Correlation with Virus Transmission to the Fetus. J. Infect. Dis. 2007, 195, 1062–1070.
- Fornara, C.; Furione, M.; Arossa, A.; Gerna, G.; Lilleri, D. Comparative magnitude and kinetics of human cytomegalovirus-specific CD4+ and CD8+ T-cell responses in pregnant women with primary vs. remote infection and in transmitting vs. non-transmitting mothers: Its utility for dating primary infection in pre. J. Med. Virol. 2016, 88, 1238–1246.
- Tabata, T.; Petitt, M.; Fang-Hoover, J.; Freed, D.C.; Li, F.; An, Z.; Wang, D.; Fu, T.-M.; Pereira, L. Neutralizing Monoclonal Antibodies Reduce Human Cytomegalovirus Infection and Spread in Developing Placentas. Vaccines 2019, 7, 135.
- Fouts, A.E.; Chan, P.; Stephan, J.-P.; Vandlen, R.; Feierbach, B. Antibodies against the gH/gL/UL128/UL130/UL131 Complex Comprise the Majority of the Anti-Cytomegalovirus (Anti-CMV) Neutralizing Antibody Response in CMV Hyperimmune Globulin. J. Virol. 2012, 86, 7444–7447.
- Freed, D.C.; Tang, Q.; Tang, A.; Li, F.; He, X.; Huang, Z.; Meng, W.; Xia, L.; Finnefrock, A.C.; Durr, E.; et al. Pentameric complex of viral glycoprotein H is the primary target for potent neutralization by a human cytomegalovirus vaccine. Proc. Natl. Acad. Sci. USA 2013, 110, E4997–E5005.
- Ciferri, C.; Chandramouli, S.; Leitner, A.; Donnarumma, D.; Cianfrocco, M.A.; Gerrein, R.; Friedrich, K.; Aggarwal, Y.; Palladino, G.; Aebersold, R.; et al. Antigenic Characterization of the HCMV gH/gL/gO and Pentamer Cell Entry Complexes Reveals Binding Sites for Potently Neutralizing Human Antibodies. PLoS Pathog. 2015, 11, e1005230.
- Chauhan, V.; Singh, M.P. Immuno-informatics approach to design a multi-epitope vaccine to combat cytomegalovirus infection. Eur. J. Pharm. Sci. 2020, 147, 105279.