Human endogenous retroviruses (HERVs) are ancient retroviral DNA sequences established into germline. They contain regulatory elements and encoded proteins few of which may provide benefits to hosts when co-opted as cellular genes. Their tight regulation is mainly achieved by epigenetic mechanisms, which can be altered by environmental factors, e.g., viral infections, leading to HERV activation. This review summarizes the recent advances on the epigenetic mechanisms controlling HERV expression and the pathogenic effects triggered by HERV de-repression leading to neurological diseases, inflammatory processes and neurodegeneration.
- amyotrophic lateral sclerosis
- epigenetics
- HERV-K
- HERV-W
- monoclonal antibody
- multiple sclerosis
- neurodegeneration
- temelimab
1. Introduction
HERVs are remnants of ancient exogenous viral infections that established the Vertebrates germline millions of years ago
HERVs are remnants of ancient exogenous viral infections that established the Vertebrates germline millions of years ago
. They classify into different families named after the amino acid one-letter code of the aminoacyl-tRNA used to prime their retrotranscription
[5]
. HERVs typically encompass three main proviral genes:
pol
,
gag
and
env,
flanked by two LTR sequences. The
gag
gene encodes structural proteins; the
pol
gene, a reverse transcriptase, a protease, and an integrase; and the
env
gene, an envelope protein. Since their introduction into the germline, some HERVs have accumulated mutations in their proviral genes, making them replication-defective because of frameshifts, the appearance of early stop codons or deletions/insertions of sequences. The homologous recombination between both LTR sequences has also led to a complete loss of proviral genes in some cases, leaving the HERV represented by its LTR only
. Through these mechanisms, evolution has left copies of TEs with variable genomic contents in the human population.
HERVs have occasionally provided their hosts with beneficial effects. They contribute to genomic variability, provide additional gene regulatory elements, such as alternative promoters or alternative splice sites
, or rule important physiological processes. HERVs also participate in transcriptional regulation with noncoding RNAs
or in the expression of “domesticated” viral proteins
. Nonetheless, if uncontrolled and/or abnormally activated, HERVs can lead to detrimental effects in their host organisms by altering gene expression profiles, expressing pathogenic nucleic acids/proteins or even inducing deleterious mutations
2. Epigenetic Control of HERVs
The expression of HERVs can be controlled at different levels, but the most important are the epigenetic regulatory mechanisms
(
). While most HERVs are constitutively repressed in the human genome, some co-opted elements must become activated in a limited way, at specific time frames and in particular cell types
. To achieve this complex context-specific regulation, some HERVs harbor binding sequences for KRAB-containing zinc finger proteins (KRAB-ZFPs). Interestingly, the number of KRAB-ZFPs correlates with the LTR transposon load across species
. In fact, these transcriptional repressors can benefit the host via the domestication of TEs, including HERVs
.
KRAB-ZFPs are comprised of an array of C2H2 zinc fingers, which confers them with specificity to bind the DNA sequences lying within HERV elements, and a Krüppel-associated box (KRAB) that recruits the corepressor KRAB-associated protein 1 (KAP1), also known as TRIM28
. KAP1 serves as a scaffold for the assembly of heterochromatin epigenetic modifiers like the histone methyl transferase SETDB1 (also known as ESET)
, the heterochromatin protein 1 (HP1)
or DNA methyl transferases (DNMTS)
(
, number 1). Three molecules of KAP1 bind to the KRAB domain of the KRAB-ZFPs through their N-terminal RING-B box-coiled coil (RBCC) region, while the C-terminal PDH domain of KAP1 functions as an intramolecular E3 ligase that sumoylates several lysines in its bromodomain. Sumoylation is a post-translational modification that consists of the covalent attachment of a Small Ubiquitin-like Modifier (SUMO) to a lysine residue. KAP1 bromodomain sumoylation enhances its corepressor activity and enables its interaction with SETDB1
. Furthermore, it enhances SETDB1 activity
(
, number 1). When SETDB1 interacts with KAP1, it methylates histone H3 at lysine 9 (H3K9me) through its C-terminal ubiquitinated SET domain
. This mechanism leads to the repression of a subset of HERVs where the H3K9me heterochromatin has not yet been established, as shown by the activation of a larger group of HERVs in SETDB1 knockouts as compared to KAP1 knockouts
. Furthermore, KAP1-mediated silencing is especially important for brain development. Its depletion in neural progenitor cells aberrantly activates ERV expression while having no impact on adult neural cells
.
Also, SETDB1 can directly bind, in a KAP1-independent way, to H3K9me1/K14ac or H3K9me2/K14ac histone tails through its N-terminal triple Tudor domain (TTD) to deacetylate K14 via SETDB1-associated histone deacetylases (HDACs) and further methylate K9 to achieve a trimethylated (H3K9me3) state
(
, number 2). SETDB1 takes part in silencing retroelements in differentiated cells, including B cells
and brain tissue
, as well as innate immune genes
.
The introduction of H3K9me3 by SETDB1 also allows the binding of HP1 to the genome, where it interacts with KAP1 as well
(
, number 1). HP1 proteins prevent transcription factor binding and help maintain the heterochromatic structure
. Indeed, the HP1 isoform α may be required to repress the expression of both immune-related genes and HERVs
. Besides SETDB1 and HP1, KAP1 also recruits other repressor proteins, such as the NuRD HDAC complex, which removes transcription-promoting histone acetylation
(
, number 1). Moreover, since the axis KRAB/KAP1/SETDB1 localizes to restricted regions within HERVs, and H3K9me3 marks must spread out all over HERV sequences to ensure their silencing, KAP1 may form a complex with HP1 and histone methyltransferases all over the HERV region to mediate the spreading of heterochromatin
.
Although some studies have highlighted the importance of histone post-translational modifications (PTMs) in regulating the HERV expression in terminally differentiated cells
, DNA methylation seems to also play a relevant role in achieving their regulation
. Furthermore, the impact of DNA methylation-dependent mechanisms on HERVs expression may vary according to cell type and the evolutionary age of the element. For example, histone PTMs mediated by SETDB1, and not DNA methylation, are essential for HERV repression in neural progenitor cells
and B cells
. Additionally, younger LTRs are found to be suppressed by DNA methylation, while older ones are mainly silenced by histone PTMs
. By contrast, both mechanisms seem to play important roles in embryonic stem cells
.
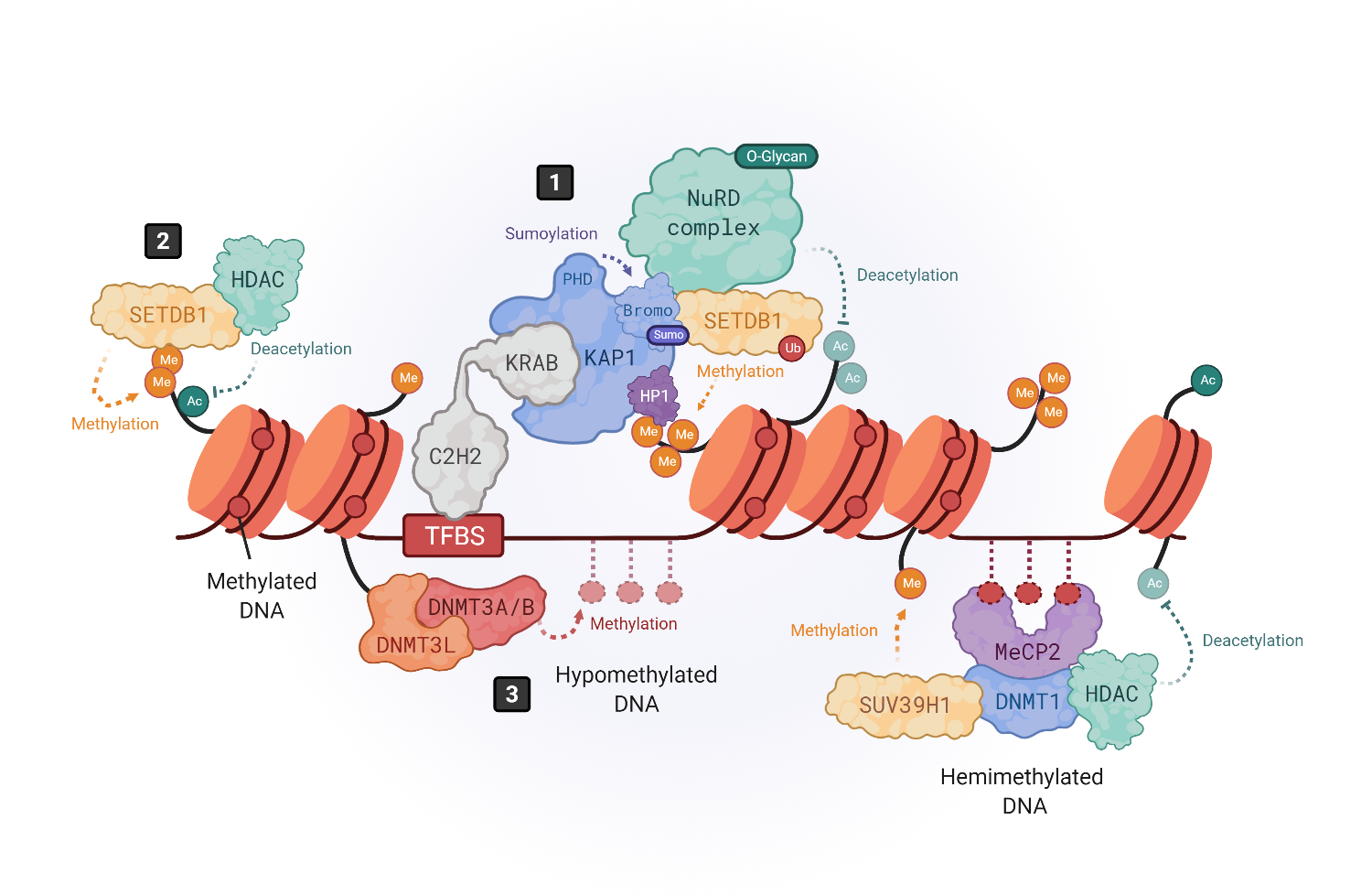
Epigenetic silencing mechanisms of HERVs. The expression of HERVs is mainly controlled by DNA methylation and histone tail modifications. Histone modifications are driven by KRAB-containing zinc finger proteins (KRAB-ZFPs), which bind to transcription factor binding site (TFBS) within HERV elements, recruiting the corepressor KRAB-associated protein 1 (KAP1), also known as TRIM28 (
)
. KAP1, in turn, serves as a scaffold molecule for the assembly of heterochromatin modifiers like the histone methyltransferase SETDB1 (also known as ESET)
, the heterochromatin protein 1 (HP1)
or the NuRD histone deacetylase complex
. Moreover, SETDB1 can also bind in a KAP1-independent way to H3K9me/K14ac marks to drive histone methylation (
)
. DNA methylation is driven by DNA methyltransferases (DNMTs). DNMT3L binds to histone 3 tails and recruits DNMT3A and DNMT3B to establish de novo methylation in hypomethylated DNA (
), whereas hemimethylated DNA is bound by methyl-binding proteins like MeCP2, which recruits repressor complexes such as DNMT1, histone deacetylases (HDACs) and methyltransferases like SUV39H1 to stabilize the chromatin structure repressing the gene expression (
)
.
DNA methylation is a modification carried out by DNA methyltransferases (DNMTs) that consists on the transfer of a methyl group onto carbon 5 (C5) of cytosines to form 5-methylcytosine, thus recruiting methyl-CpG-binding proteins (MBP) and/or preventing the binding of transcription factors
. It has been shown that the brain contains some of the highest DNA methylation levels in the body and that MBPs are important for normal neuronal functions, being highly and specifically expressed in the brain
. DNA methylation can be promoted at specific regions of the genome through transcription factor or RNA interference (RNAi)-mediated mechanisms
or simply spread across susceptible genomic regions. In hypomethylated DNA, DNMT3L binds to histone 3 tails and recruits DNMT3A and DNMT3B to establish de novo methylation
(
, number 3).
Additionally, DNMT3A/B can bind hypomethylated DNA regions, unprotected by transcription factors. When DNA is hemimethylated, MBPs like MeCP2 bind to DNA and recruit various repressor complexes such as DNMT1, or H3K9 methyltransferases like SUV39H1, to achieve DNA and histone methylation
, and histone deacetylases (HDAC) to remove activating histone modifications
(
, number 4). Thus, DNA methylation and histone modifications work closely to induce a heterochromatic-repressive state. In fact, one study demonstrated that DNMT3L facilitates the binding of KAP1 to various repressive epigenetic modifiers
.
In addition to DNA methylation and histone modifications, transcription factor PTMs also play an important role in HERV silencing. As mentioned earlier, KAP1 sumoylation enhances its recruitment to retroelements but, also, the recruitment of SETDB1 to mediate H3K9 trimethylation
(
, number 1). In addition, the inhibitory activity of KAP1-associated repressive chromatin factors, such as the HDAC1 of the NuRD complex
, is enhanced by methylation-dependent O-linked glycosylation, a PTM introduced by the O-linked β-
-acetylglucosamine transferase (OGT)
(
, number 1). On the other hand, HERVs can also be post-transcriptionally regulated by RNA-binding proteins, such as the RNA-Binding Motif Protein 4 (RBM4)
. RBM4 regulates HERV-K (HML-2) and the HERV-H transcript levels by binding at CGG consensus elements present on 5′ and 3′ LTRs. However, the mechanism by which RBM4 mediates HERV downregulation remains unknown. Another post-transcriptional regulatory mechanism is m6A RNA methylation, which reduces the half-life of ERV mRNAs, protecting cell integrity, as shown in mouse cells
. Thus, a combination of multiple heterochromatin modifiers, their PTMs, and the post-transcriptional regulation of HERV transcripts, may prevent the ectopic and/or pathogenic expression of their encoded nucleic acid sequences and proteins that could be sensed by the immune system, triggering inflammatory responses
.
23. Transactivation of HERVs
Despite their tightly controlled regulation, pathogenic HERV proteins have been detected in chronic inflammatory or degenerative diseases of the nervous system, such as multiple sclerosis (MS)
, amyotrophic lateral sclerosis (ALS) [
] or schizophrenia
. One of the main families being related to these neurological diseases is the HERV-W
.
The HERV-W family is present in the human genome in multiple copies, most of them being defective and replication-incompetent
. The only known HERV-W family member encoding a complete
gene that transcribes and translates into a complete Env protein is ERVWE-1, a replication-incompetent HERV-W element located on chromosome 7q21.2
that encodes Syncytin-1, a protein found only intracellularly or on the plasma membrane and is, therefore, cell-associated. Syncytin-1 plays an important role during early pregnancy by facilitating trophoblastic cell fusion to allow embryo implantation
. By contrast, MS-associated retrovirus (MSRV), now referred to as pHERV-W ENV, have been found extracellularly, in the cerebrospinal fluid (CSF) of MS patients, as genomic RNA assembled into proviral particles
. The origin of HERV-W/MSRV remains uncertain, since any known HERV-W family member could, in principle, yield viral particles, and no human genome locus encoding pHERV-W ENV has yet been found in the human genome. Thus, it has been suggested that it originates from the complementation of different HERV-W elements, by reversing the stop codon in the Xq22.3 locus
, from a non-ubiquitous and/or unfixed copy
or could even be an exogenous member of the HERV-W family
. Interestingly, and likewise for all HERV-W sequences, the pHERV-W ENV length only differs from Syncytin-1 by the presence of four additional amino acids. Despite a reported identity of 81%
, several important differences between these two proteins have been detected. Firstly, Syncytin-1 is a membrane-associated protein, while pHERV-W ENV can be membrane-associated within viral particles or as a hexameric-soluble form, although monomer and trimers have also been described
. Secondly, HERV-W/MSRV is the main family member associated with chronic inflammatory diseases, though ectopic pathogenic Syncytin-1 expression has been reported in schizophrenia
.
The activation of the HERV-W sequences may derive from dysregulation of the epigenetic mechanisms controlling their expression, since the inhibition of DNMTs and HDACs leads to HERV reactivation
. As epigenetic mechanisms can be influenced by environmental stimuli, it seems possible that some environmental factors may distort HERV silencing mechanisms, reactivating their expression and, thereby, inducing or contributing to disease. In fact, some viral infections have been shown to lead to reactivation of HERVs and, interestingly, are believed to be cofactors of HERV-associated diseases. For example,
infections are associated with a higher risk for developing MS
, while influenza seems to contribute to schizophrenia, especially when infections occur during the prenatal stages
.
Influenza virus has been shown to transactivate HERV-W elements by impairing the KRAB-ZFP/KAP1/SETDB1 axis, which plays an important role in controlling HERV expression, as described above (
, number 1). In vitro, influenza virus triggers loss of sumoylations on KAP1 and lowers the levels of H3K9 trimethylation and SETDB1 without affecting promoter DNA methylation status. This leads to de-repression of HERV-W elements like Syncytin-1
.
Besides, herpes viruses like Herpes Simplex Virus type 1 (HSV-1), Human Herpes Virus type 6 (HHV6) and Epstein-Barr Virus (EBV) are efficient at activating HERV-W elements, even on methylated promoters
. Another HERV-W activator is HHV6
. For the Epstein–Barr virus (EBV), in vitro experiments showed that the binding of the EBV major envelope protein gp350 activates the transcription of HERV-W/MSRV
and Syncytin-1 in peripheral blood mononuclear cells (PBMCs) and astrocytes without requiring EBV entry
.
However, not only viral infections have the capacity for HERV activation; cytokines can also induce their expression
. This opens up the intriguing possibility that the activation of HERVs in inflammatory diseases serves to create a feedback loop amplifying abnormal immune responses when pathogenic HERVs have been epigenetically de-repressed and activated.
Like MS, no single environmental stimuli or single gene mutations have been described as the disease-causing agents for schizophrenia or ALS, but a combination of environmental factors and genetic susceptibility backgrounds have been proposed
. It is tempting to think that HERV activation may represent the common denominator to those risk factors, with particular combinations leading to the reactivation of specific HERV elements. In this hypothetical scenario, depending on the HERV(s) element(s) de-repressed and the cell type(s) involved, the resulting symptoms and, thereby, the disease triggered would be one or another.
HERV-W Related Diseases
One of the most studied pathologies linked to abnormal HERV expression is MS, an autoimmune neurodegenerative disease that affects myelin sheaths, leading to neuronal loss [65][101]. The commonly used experimental model for MS is the mouse autoimmune model EAE (Experimental Autoimmune Encephalomyelitis) [102], from which most of the knowledge about neuroinflammation and neurodegeneration derives. EAE is induced by immunization with myelin or central nervous system (CNS)-derived peptides, and the disease is mediated by activated CNS-specific CD4+ T cells in the periphery, which enter the CNS by crossing the endothelial blood–brain barrier (BBB). By contrast, neuroinflammation in MS has now been revealed to be initiated in the CNS with the activation of microglia [103][104]. Under physiological conditions, microglia protect the brain by sensing pathogens, phagocytosing cell debris and promoting remyelination. However, under MS pathological conditions, microglia become activated and secrete proinflammatory molecules that directly damage myelin sheaths and oligodendrocytes, causing inflammation and neurodegeneration [105][106] (Figure 2, number 1). This proinflammatory environment attracts peripheral monocyte-derived macrophages and dendritic cells (DC), as well as peripheral autoreactive T-lymphocytes, which enter the CNS through the BBB localized in CNS microvessels [103][107][108][109] (Figure 2, number 2). Infiltrating T-lymphocytes become activated by microglia, which also act as antigen-presenting cells (APCs), phagocytosing myelin debris and expressing major histocompatibility complexes (MHC) I and II with costimulatory molecules (Figure 2, number 3). Infiltrated monocyte-derived DCs also act as APCs, presenting myelin antigen to T cells, along with microglia [110] (Figure 2, number 3). Activated T-lymphocytes differentiate into T-helper cell-type 1 (Th1) (producing IFN-γ) and Th17 cells (producing IL-17, IL-22 and IL-21) [103][105][106][111][112] (Figure 2, number 3). The crosstalk between neuroimmune and peripheral immune systems exacerbates neuroinflammation, further activating microglia and astrocytes through cytokine production, as well as facilitating the infiltration of other immune cells, such as activated B cells, monocytes and macrophages. The release of proinflammatory cytokines, nitric oxide, reactive oxygen and nitrogen species [113], together with some other processes like phagocytosis of myelin by macrophages and antibody-dependent cytotoxicity, contribute to inflammation within the CNS, oligodendrocyte loss, and demyelination of the white and gray matter [114].
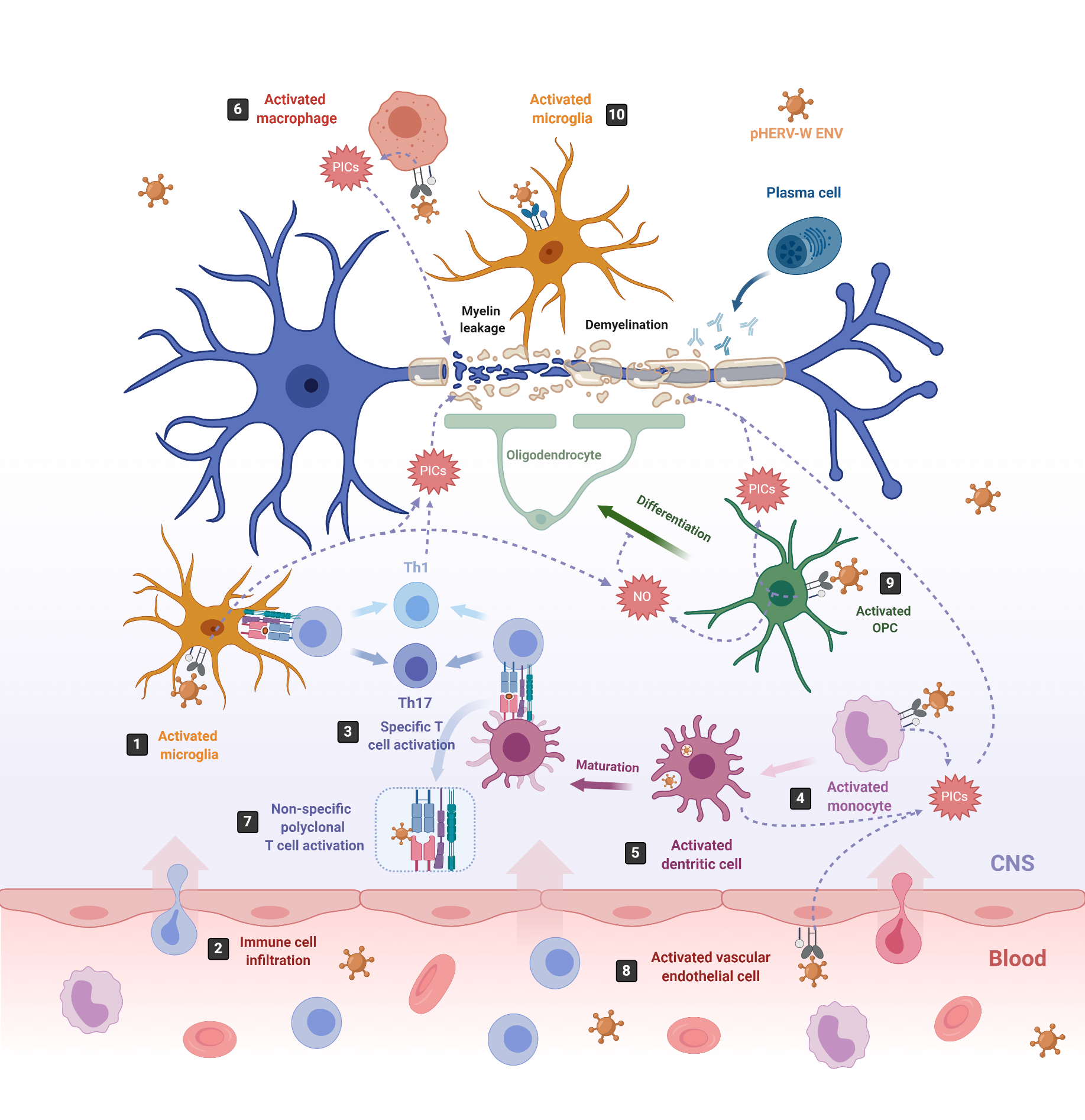
Figure 2. pHERV-W-ENV drives inflammation and neurodegeneration in MS. In MS, inflammation starts in the CNS with the activation of microglia, which release proinflammatory cytokines (PICs) that directly damage the myelin sheaths (1). Cell debris and PICs attract peripheral immune cells to the CNS (2); among them are T cells, which are then presented by APCs like microglia or infiltrating monocyte-derived dendritic cells (3) [103]. In this sense, pHERV-W ENV stimulates the innate immune system by engaging TLR4/CD14 receptors in microglia (1) monocytes (4), dendritic cells (5) and macrophages (6), inducing the release of PICs. In addition, it stimulates specific T-cell activation (3) and abnormal polyclonal T-cell activation (7) [19][18], acting as a superantigen. Furthermore, pHERV-W ENV activates vascular endothelial cells (8), making the migration of circulating cells possible at the level of ongoing brain lesions initiated by perivascular macrophages and/or microglia [115]. On the other hand, pHERV-WENV impairs the remyelination process by inducing the release of PICs and nitrosative stress in oligodendrocyte precursor cells (OPCs) (9) and the microglia (1), inhibiting the differentiation of OPCs to oligodendrocytes and damaging the myelin sheath, ultimately leading to neuronal loss [116]. pHERV-W ENV also drives microglial cells to physically interact with myelinated axons and induces the leakage of intra-axonal and myelin proteins (10) [117].
Although neurodegeneration has traditionally been thought to be a consequence of inflammation in the CNS, this concept has been challenged by the poor efficacy of immunomodulatory treatments on preventing demyelination and axonal degeneration [115]. Now, we know that pHERV-W ENV may participate in the development of MS by triggering a potent immune response, as well as by impairing the myelin-repairing process [18][19][115][117][118][119][120], which could explain why immunomodulatory treatments failed at preventing neurodegeneration. Furthermore, pHERV-W ENV contribution to MS is supported by the observation that administered pHERV-W ENV with myelin oligodendrocyte glycoprotein (MOG) 35–55 peptide triggers EAE in mice [119].
pHERV-W ENV RNA and protein have been detected in PBMCs of MS patients [74][76][81][121], showing their highest RNA levels in natural killer (NK) cells, followed by B cells and monocytes, the last increasing their expression by differentiation to macrophages. By contrast, no ENV-encoding RNA or protein is detected in T cells [66][122]. pHERV-W ENV has also been detected in astrocytes [66][75], infiltrated macrophages and activated microglia in the brains [123][121][124] of MS patients, and its soluble form in their serum, plasma, and CSF [65][76][125][126][127][128]. Indeed, the presence of HERV-W RNA in CSF is considered as a negative prognostic marker of MS. Its load increases with MS duration and parallels the clinical stages [126][127][128][129][130].
The SU domain of the pHERV-W ENV, either associated to viral particles or in its soluble form has been shown to trigger proinflammatory responses in vitro, in human PBMCs and dendritic cells (DCs) [19] and, in vivo, in humanized severe combined immunodeficiency (SCID) mice, which die from brain hemorrhages with an overexpression of TNF-α after pHERVW-ENV injection [18][131]. The inflammatory reaction induced by pHERV-W ENV is mediated by the activation of Toll-like receptor (TLR) 4 and its coreceptor CD14 [19][119]. TLRs are expressed by different cell types, such as B cells [132], macrophages, monocytes, DCs [133], oligodendrocytes [134], astrocytes [135] or microglia [136], among other, where they mediate part of the innate immune response against invading pathogens [137][138].
On the one hand, pHERV-W ENV activates monocytes, DCs and macrophages (Figure 2, number 4–6). Activated monocytes produce proinflammatory cytokines, including TNFα, IL-1β, IL-6 and IL-12p40 (Figure 2, number 4). Interestingly, the levels of IL-6 and IL-12p40 induced in cultured PBMCs directly correlate with the MS disease severity [19]. Activated DCs not only release the proinflammatory cytokines IL-6, TNFα, IL-12p40 and IL-12p70 but, also, upregulate antigen presentation, activating antigen-specific T cells. The release of IL-12 by DCs activates naïve T cells and promotes their differentiation into IFN γ-secreting Th1 cells [19] (Figure 2, number 3). In turn, released INF-γ and TNF-α upregulate pHERV-W ENV secretion by PBMCs [81]. Thus, pHERV-W ENV not only activates the innate immune system but, also, the adaptative immune system. Moreover, it also acts like a superantigen [18], stimulating abnormal polyclonal T-cell activation through its binding to Vβ chains, and the expansion of T-cell-receptor β-chain (TCR Vβ) cells, leading to nonspecific oligoclonal Vβ+T cell activation [18][139] (Figure 2, number 7).
In addition, pHERV-W ENV activates vascular endothelial cells within the BBB, leading to the release of proinflammatory cytokines IL-6 and IL-8 and to the overexpression of ICAM-1, thus enhancing the migration of circulating cells towards the brain (Figure 2, number 8) [115]. In the brain, the pHERV-W ENV-SU domain triggers CNS innate immunity through TLR4 binding, activating microglia and perivascular macrophages, which release proinflammatory cytokines, leading to neuroinflammation and neurodegeneration [140] (Figure 2, number 1). Additionally, pHERV-W ENV produced by the surrounding cells like microglia or macrophages [74] engages with, and activates, TLR4 receptors present on the brain’s resident oligodendrocyte precursor cells (OPCs). The OPCs’ role is to migrate to the lesion sites where oligodendrocytes are depleted, and differentiate into mature oligodendrocytes to remyelinate axons after an injury [141]. After migration, OPCs transiently express TLR4 receptors, which become downregulated during the OPCs’ differentiation process to oligodendrocytes [116]. TLR4 activation does not affect OPC survival; however, it induces the release of proinflammatory cytokines such as TNF-α, IL-1β and IL-6, as well as the overexpression of inducible nitric oxide synthase (iNOS), leading to an increase in nitric oxide (NO) levels that triggers nitrosative stress, which, in turn, affects the myelin protein expression and reduces OPCs’ differentiation capacity [116] (Figure 2, number 9). NO and TNF-α are well-established mediators of axonal injury and demyelination [142][143]. Interestingly, TLR4 activation in OPCs seems to be sensitive to the cyclin-dependent kinase inhibitor p57kip2, which participates in controlling TLR4’s surface expression [118].
On the other hand, pHERV-W ENV induces a degenerative phenotype in microglial cells characterized by the expression of the same set of proinflammatory cytokines detected on pHERV-W ENV-activated OPCs (TNF-α, IL-1β and IL-6) and by the overexpression of iNOS [117] (Figure 2, number 1). While reducing anti-inflammatory and neuroprotective proteins and downregulating the expression of genes involved in myelin debris phagocytosis, it affects neurorepair and inhibits OPC differentiation. Furthermore, the pHERV-W ENV protein stimulates microglia proliferation and drives physical interaction between microglial cells and myelinated axons to induce leakage of intra-axonal and myelin proteins [117] (Figure 2, number 10). Of note, the multifaceted effects of pHERV-W ENV are often due to its ability to strongly bind TLR4 receptors on expressing cells, thereby activating underlying signaling pathways and leading to various effects with a proinflammatory common point in different cells [144].
Furthermore, pHERV-W-ENV inflammatory and demyelinating consequences can also be observed in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), a rare immune disease of the peripheral nervous system (PNS) [120].In these patients, pHERV-W ENV is found in PBMCs and peripheral nerve lesions, especially in Human Schwann Cells (HSCs), in which it exerts pathogenic effects by TLR4 engagement, inducing the release of proinflammatory IL-6 and the macrophage and T-cell chemoattractant CXCL10 [145], triggering activation of the innate immune system.
Another HERV-W-related disease is schizophrenia [146][147], a psychiatric disorder that highly impacts patients quality of life. In reference to the mechanisms underlying this disease, several are the hypotheses raised, including the disturbance of the dopaminergic neurotransmission or its relationship with neurodevelopmental problems. However, the idea that autoimmune processes within the nervous system could be playing an important role in its pathophysiology has been gaining more and more acceptance [148]. Some studies have shown the elevated expression of TLRs [149] and abnormal expression of proinflammatory cytokines, like IL-6, IL-8 and the C-reactive protein (CRP), in the serum of these patients [150][151]. CRP is released to blood under inflammatory conditions, and its overexpression supports that immune mechanisms play a role in schizophrenia symptoms beyond cognitive decline [152]. Interestingly, the levels of HERV-W mRNA were found to associate with a proinflammatory phenotype in these patients [153].
HERV-W expression is particularly associated with recent-onset schizophrenia [68][146][154][155]. Both HERV-W/MSRV and Syncytin-1 were reported to be increased in schizophrenia [84][156], and HERV-W/MSRV was shown to be unequally represented in the genome of an affected and its nonaffected monozygotic twin [157]. HERV-W GAG and ENV RNA or protein have been found in the circulating blood of these patients [147][154][155][156][158], correlating GAG protein levels with disease severity [158] and Syncytin-1 or (MSRV) pHERV-W ENV with CRP levels [155], providing a link between HERV-W envelopes and the status of systemic inflammation in these patients [84][147][155]. Activation of CRP in microglia and astrocytes, and the consequent inflammation, can be triggered by Syncytin-1 through direct interaction with TLR3 receptors [84] and IL-6 release. Furthermore, HERV-W presence has also been detected in CSF [68] and in cortical tissue [159], indicating a pathogenic role in the brain. In fact, HERV-W/MSRV induces activation of the Brain-derived neurotrophic factor (BDNF) and Dopamine receptor D3 (DRD3) genes [156], both implicated in providing a higher risk for suffering schizophrenia [160]. Thus, HERV-W Syncytin-1 and MSRV may determine and/or promote the development of schizophrenia. In this regard, it should be mentioned that, by combining single-molecule tracking, calcium imaging and behavioral approaches, Johansson et al. [70] showed that the mechanism used by HERV-W-ENV to alter glutamate synapse maturation and generate behavioral deficits depends on the N-methyl-d-aspartate receptor (NMDAR) organization and cytokine-dependent changes, further supporting an etiological role for this HERV product in the development of psychosis.
References
- Löwer, R.; Löwer, J.; Kurth, R. The viruses in all of us: Characteristics and biological significance of human endogenous retrovirus sequences. Proc. Natl. Acad. Sci. USA 1996, 93, 5177–5184, doi:10.1073/pnas.93.11.5177.
- Sverdlov, E.D. Retroviruses and primate evolution. Bioessays 2000, 22, 161–171, doi:10.1002/(SICI)1521-1878(200002)22:2<161::AID-BIES7>3.0.CO;2-X.
- Bannert, N.; Kurth, R. The evolutionary dynamics of human endogenous retroviral families. Annu. Rev. Genom. Hum. Genet. 2006, 7, 149–173, doi:10.1146/annurev.genom.7.080505.115700.
- Johnson, W.E. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat. Rev. Microbiol. 2019, 17, 355–370, doi:10.1038/s41579-019-0189-2.
- Griffiths, D.J. Endogenous retroviruses in the human genome sequence. Genome Biol. 2001, 2, 1–5, doi:10.1186/gb-2001-2-6-reviews1017.
- Saleh, A.; Macia, A.; Muotri, A.R. Transposable elements, inflammation, and neurological disease. Front. Neurol. 2019, 10, 894, doi:10.3389/fneur.2019.00894.
- Belshaw, R.; Watson, J.; Katzourakis, A.; Howe, A.; Woolven-Allen, J.; Burt, A.; Tristem, M. Rate of recombinational deletion among human endogenous retroviruses. J. Virol. 2007, 81, 9437–9442, doi:10.1128/JVI.02216-06.
- Medstrand, P.; Landry, J.R.; Mager, D.L. Long terminal repeats are used as alternative promoters for the endothelin B receptor and apolipoprotein C-I genes in humans. J. Biol. Chem. 2001, 276, 1896–1903, doi:10.1074/jbc.M006557200.
- Jern, P.; Coffin, J.M. Effects of retroviruses on host genome function. Annu. Rev. Genet. 2008, 42, 709–732, doi:10.1146/annurev.genet.42.110807.091501
- Suntsova, M.; Garazha, A.; Ivanova, A.; Kaminsky, D.; Zhavoronkov, A.; Buzdin, A. Molecular functions of human en-dogenous retroviruses in health and disease. Cell Mol. Life Sci. 2015, 72, 3653–3675.
- Ito, J.; Sugimoto, R.; Nakaoka, H.; Yamada, S.; Kimura, T.; Hayano, T.; Inoue, I. Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLoS Genet. 2017, 13, e1006883, doi:10.1371/journal.pgen.1006883
- Hu, T.; Pi, W.; Zhu, X.; Yu, M.; Ha, H.; Shi, H.; Choi, J.-H.; Tuan, D. Long non-coding RNAs transcribed by ERV-9 LTR retrotransposon act in cis to modulate long-range LTR enhancer function. Nucleic Acids Res. 2017, 45, 4479–4492, doi:10.1093/nar/gkx055.
- Wilson, K.D.; Ameen, M.; Guo, H.; Abilez, O.J.; Tian, L.; Mumbach, M.R.; Diecke, S.; Qin, X.; Liu, Y.; Yang, H.; et al. En-dogenous Retrovirus-Derived lncRNA BANCR Promotes Cardiomyocyte Migration in Humans and Non-human Primates. Dev. Cell 2020 54, 694–709, doi:10.1016/j.devcel.2020.07.006.
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789.
- Mallet, F.; Bouton, O.; Prudhomme, S.; Cheynet, V.; Oriol, G.; Bonnaud, B.; Lucotte, G.; Duret, L.; Mandrand, B. The en-dogenous retroviral locus ERVWE1 is a bona fide gene involved in hominoid placental physiology. Proc. Natl. Acad. Sci. USA 2004, 101, 1731–1736, doi:10.1073/pnas.0305763101.
- Larsson, E.; Andersson, G. Beneficial role of human endogenous retroviruses: Facts and hypotheses. Scand. J. Immunol. 1998, 48, 329–338, doi:10.1046/j.1365-3083.1998.00428.x.
- Mangeney, M.; Renard, M.; Schlecht-Louf, G.; Bouallaga, I.; Heidmann, O.; Letzelter, C.; Richaud, A.; Ducos, B.; Heidmann, T. Placental syncytins: Genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 20534–20539, doi:10.1073/pnas.0707873105
- Perron, H.; Jouvin-Marche, E.; Michel, M.; Ounanian-Paraz, A.; Camelo, S.; Dumon, A.; Jolivet-Reynaud, C.; Marcel, F.; Souillet, Y.; Borel, E; et al. Multiple sclerosis retrovirus particles and recombinant envelope trigger an abnormal immune response in vitro, by inducing polyclonal Vbeta16 T-lymphocyte activation. Virology 2001, 287, 321–332, doi:10.1006/viro.2001.1045.
- Rolland, A.; Jouvin-Marche, E.; Viret, C.; Faure, M.; Perron, H.; Marche, P.N. The envelope protein of a human endogenous retrovirus-W family activates innate immunity through CD14/TLR4 and promotes Th1-like responses. J. Immunol. 2006, 176, 7636–7644, doi:10.4049/jimmunol.176.12.7636.
- Hurst, T.P.; Magiorkinis, G. Epigenetic control of human endogenous retrovirus expression: Focus on regulation of long-terminal repeats (LTRs). Viruses 2017, 9, 130, doi:10.3390/v9060130.
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell Mol. Life Sci. 2017, 74, 2055–2065, doi:10.1007/s00018-017-2454-8.
- Turelli, P.; Playfoot, C.; Grun, D.; Raclot, C.; Pontis, J.; Coudray, A.; Thorball, C.; Duc, J.; Pankevich, E.V.; Deplancke, B.; et al. Primate-restricted KRAB zinc finger proteins and target retrotransposons control gene expression in human neurons. Sci. Adv. 2020, 6, eaba3200, doi:10.1126/sciadv.aba3200.
- Thomas, J.H.; Schneider, S. Coevolution of retroelements and tandem zinc finger genes. Genome Res. 2011, 21, 1800–1812, doi:10.1101/gr.121749.111.
- Ecco, G.; Imbeault, M.; Trono, D. A tale of domestication: The endovirome, its polydactyl controllers and the spe-cies-specificity of human biology. Development 2020, 144, 2719–2729, doi:10.1242/dev.132605.
- Iyengar, S.; Farnham, P.J. KAP1 protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011, 286, 26267–26276, doi:10.1074/jbc.R111.252569.
- Ecco, G.; Imbeault, M.; Trono, D. KRAB zinc finger proteins. Development 2017, 144, 2719–2729, doi:10.1242/dev.132605.
- Stoll, G.A.; Oda, S.-I.; Chong, Z.-S.; Yu, M.; McLaughlin, S.H.; Modis, Y. Structure of KAP1 tripartite motif identifies mo-lecular interfaces required for retroelement silencing. Proc. Natl. Acad. Sci. USA 2019, 116, 15042–15051, doi:10.1073/pnas.1901318116.
- Grassi, D.A.; Jonsson, M.E.; Brattas, P.L.; Jakobsson, J. TRIM28 and the control of transposable elements in the brain. Brain Res. 2019, 1705, 43–47, doi:10.1016/j.brainres.2018.02.043.
- Tie, C.H.; Fernandes, L.; Conde, L.; Robbez-Masson, L.; Sumner, R.P.; Peacock, T.; Rodriguez-Plata, M.T.; Mickute, G.; Gifford, R.; Towers, G.J.; et al. KAP1 regulates endogenous retroviruses in adult human cells and contributes to innate immune control. EMBO Rep. 2018, 19, e45000, doi:10.15252/embr.201745000.
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., III. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932, doi:10.1101/gad.973302.
- Ryan, R.F.; Schultz, D.C.; Ayyanathan, K.; Singh, P.B.; Friedman, J.R.; Fredericks, W.J.; Rauscher, F.J., III. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: A potential role for Krüppel-associated box–zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell Biol. 1999, 19, 4366–4378, doi:10.1128/mcb.19.6.4366.
- Turelli, P.; Castro-Diaz, N.; Marzetta, F.; Kapopoulou, A.; Raclot, C.; Duc, J.; Tieng, V.; Quenneville, S.; Trono, D. Interplay of TRIM28 and DNA methylation in controlling human endogenous retroelements. Genome Res. 2014, 24, 1260–1270, doi:10.1101/gr.172833.114.
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837, doi:10.1016/j.molcel.2007.11.012.
- Ishimoto, K.; Kawamata, N.; Uchihara, Y.; Okubo, M.; Fujimoto, R.; Gotoh, E.; Kakinouchi, K.; Mizohata, E.; Hino, N.; Okada, Y.; et al. Ubiquitination of lysine 867 of the human SETDB1 protein upregulates its histone H3 lysine 9 (H3K9) methyl-transferase activity. PLoS ONE 2016, 11, e0165766, doi:10.1371/journal.pone.0165766.
- Jönsson, M.E.; Garza, R.; Sharma, Y.; Petri, R.; Södersten, E.; Johansson, J.G.; Johansson, P.A.; Atacho, D.A.; Pircs, K.; Madsen, S.; et al. Activation of endogenous retroviruses during brain development causes an inflammatory response. EMBO J. 2021, 40, e106423, doi:10.15252/embj.2020106423.
- Jurkowska, R.Z.; Qin, S.; Kungulovski, G.; Tempel, W.; Liu, Y.; Bashtrykov, P.; Stiefelmaier, J.; Jurkowski, T.P.; Kudithipudi, S.; Weirich, S.; et al. H3K14ac is linked to methylation of H3K9 by the triple Tudor domain of SETDB1. Nat. Commun. 2017, 8, doi:10.1038/s41467-017-02259-9.
- Collins, P.L.; Kyle, K.E.; Egawa, T.; Shinkai, Y.; Oltz, E.M. The histone methyltransferase SETDB1 represses endogenous and exogenous retroviruses in B lymphocytes. Proc. Natl. Acad. Sci. USA 2015, 112, 8367–8372, doi:10.1073/pnas.1422187112.
- Tan, S.L.; Nishi, M.; Ohtsuka, T.; Matsui, T.; Takemoto, K.; Kamio-Miura, A.; Aburatani H.; Shinkai, Y.; Kageyama, R. Es-sential roles of the histone methyltransferase ESET in the epigenetic control of neural progenitor cells during development. Development 2012, 139, 3806–3816, doi:10.1242/dev.082198.
- Machida, S.; Takizawa, Y.; Ishimaru, M.; Sugita, Y.; Sekine, S.; Nakayama, J-I.; Wolf, M.; Kurumizaka, H. Structural basis of heterochromatin formation by human HP1. Mol. Cell. 2018, 69, 385–397, doi:10.1016/j.molcel.2017.12.011.
- Sharma, P.; Azebi, S.; England, P.; Christensen, T.; Møller-Larsen, A.; Petersen, T.; Batsché, E.; Muchardt, C. Citrullination of histone H3 interferes with HP1-mediated transcriptional repression. PLoS Genet. 2012, 8, e1002934, doi:10.1371/journal.pgen.1002934.
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J., III. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: The PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev. 2001, 15, 428–443, doi:10.1101/gad.869501.
- Stewart, M.D.; Li, J.; Wong, J. Relationship between histone H3 lysine 9 methylation, transcription repression, and hetero-chromatin protein 1 recruitment. Mol. Cell Biol. 2005, 25, 2525–2538, doi:10.1128/MCB.25.7.2525-2538.2005.
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010, 6, e1000869, doi:10.1371/journal.pgen.1000869.
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38.
- Liang, G.; Chan, M.F.; Tomigahara, Y.; Tsai, Y.C.; Gonzales, F.A.; Li E.; Laird, P.W.; Jones, P.A. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell Biol. 2002, 22, 480–491, doi:10.1128/MCB.22.2.480-491.2002.
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell. 2011, 8, 676–687, doi:10.1016/j.stem.2011.04.004.
- Fasching, L.; Kapopoulou, A.; Sachdeva, R.; Petri, R.; Jönsson, M.E.; Männe, C.; Turelli, P.; Jern, P.; Cammas, F.; Trono, D.; et al. TRIM28 represses transcription of endogenous retroviruses in neural progenitor cells. Cell Rep. 2015, 10, 20–28, doi:10.1016/j.celrep.2014.12.004.
- Ohtani, H.; Liu, M.; Zhou, W.; Liang, G.; Jones, P.A. Switching roles for DNA and histone methylation depend on evolu-tionary ages of human endogenous retroviruses. Genome Res. 2018, 28, 1147–1157, doi:10.1101/gr.234229.118.
- Sharif, J.; Endo, T.A.; Nakayama, M.; Karimi, M.M.; Shimada, M.; Katsuyama, K.; Goyal, P.; Brind’Amour, J.; Sun, M-A.; Sun, Z.; et al. Activation of endogenous retroviruses in dnmt1(−/−) ESCs involves disruption of SETDB1-mediated repression by NP95 binding to hemimethylated DNA. Cell Stem Cell. 2016, 19, 81–94 doi:10.1016/j.stem.2016.03.013.
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188, doi:10.1038/13810.
- Morris, K.V.; Chan, S.W-L.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292, doi:10.1126/science.1101372.
- Ooi, S.K.T.; Qiu, C.; Bernstein, E.; Li, K.; Jia, D.; Yang, Z.; Erdjument-Bromage, H.; Tempst, P.; Lin, S-P.; Allis, C.D.; et al. DNMT3L connects unmethylated lysine 4 of histone H3 to de novo methylation of DNA. Nature 2007, 448, 714–717, doi:10.1038/nature05987.
- Zhang, Y.; Jurkowska, R.; Soeroes, S.; Rajavelu, A.; Dhayalan, A.; Bock, I.; Rathert, P.; Brandt, O.; Reinhardt, R.; Fischle, W.; et al. Chromatin methylation activity of Dnmt3a and Dnmt3a/3L is guided by interaction of the ADD domain with the histone H3 tail. Nucleic Acids Res. 2010, 38, 4246–4253, doi:10.1093/nar/gkq147.
- Rea, S.; Eisenhaber, F.; O’Carroll, D.; Strahl, B.D.; Sun, Z.W.; Schmid, M.; Opravil, S.; Mechtler, K.; Ponting, C.P.; Allis, D.; et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000, 406, 593–599, doi:10.1038/35020506.
- Fuks, F.; Hurd, P.J.; Deplus, R.; Kouzarides, T. The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 2003, 31, 2305–2312, doi:10.1093/nar/gkg332.
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 2003, 278, 4035–4040, doi:10.1074/jbc.M210256200.
- Kimura, H.; Shiota, K. Methyl-CpG-binding protein, MeCP2, is a target molecule for maintenance DNA methyltransferase, Dnmt1. J. Biol. Chem. 2003, 278, 4806–4812, doi:10.1074/jbc.M209923200.
- Fuks, F.; Burgers, W.A.; Brehm, A.; Hughes-Davies, L.; Kouzarides, T. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat. Genet. 2000, 24, 88–91, doi:10.1038/71750.
- Kao, T.-H.; Liao, H.-F.; Wolf, D.; Tai, K.-Y.; Chuang, C.-Y.; Lee, H.-S.; Kuo, H.-C.; Hata, K.; Zhang, X.; Cheng, X.; et al. Ectopic DNMT3L triggers assembly of a repressive complex for retroviral silencing in somatic cells. J. Virol. 2014, 88, 10680–10695, doi:10.1128/JVI.01176-14.
- Zhu, G.; Tao, T.; Zhang, D.; Liu, X.; Qiu, H.; Han, L.; Xu, Z.; Xiao, Y.; Cheng, C.; Shen, A. O-GlcNAcylation of histone deacetylases 1 in hepatocellular carcinoma promotes cancer progression. Glycobiology 2016, 26, 820–833, doi:10.1093/glycob/cww025.
- Gao, J.; Yang, Y.; Qiu, R.; Zhang, K.; Teng, X.; Liu, R.; Wang, Y. Proteomic analysis of the OGT interactome: Novel links to epithelial-mesenchymal transition and metastasis of cervical cancer. Carcinogenesis 2018, 39, 1222–1234, doi:10.1093/carcin/bgy097.
- Boulard, M.; Rucli, S.; Edwards, J.R.; Bestor, T.H. Methylation-directed glycosylation of chromatin factors represses re-trotransposon promoters. Proc. Natl. Acad. Sci. USA 2020, 117, 14292–14298, doi:10.1073/pnas.1912074117.
- Foroushani, A.K.; Chim, B.; Wong, M.; Rastegar, A.; Barbian, K.; Martens, C.; Hafner, M.; Muljo, S.A. Post-transcriptional regulation of human endogenous retroviruses by RNA-Binding Motif Protein 4, RBM4. Proc. Natl. Acad. Sci. USA 2020, 117, 26520–26430 doi:10.1073/pnas.2005237117.
- Chelmicki, T.; Roger, E.; Teissandier, A.; Dura, M.; Bonneville, L.; Rucli, S.; Dossin, F.; Fouassier, C.; Lameiras, S.; Bourc’his, D. m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 2021, 591, 312–316, doi:10.1038/s41586-020-03135-1.
- Perron, H.; Garson, J.A.; Bedin, F.; Beseme, F.; Paranhos-Baccala, G.; Komurian-Pradel, F.; Mallet, F.; Tuke, P.W.; Voisset, C.; Blond, J.L.; et al. Molecular identification of a novel retrovirus repeatedly isolated from patients with multiple sclerosis.. Proc. Natl. Acad. Sci. USA 1997, 94, 7583–7588, doi:10.1073/pnas.94.14.7583.
- Mameli, G.; Poddighe, L.; Mei, A.; Uleri, E.; Sotgiu, S.; Serra, C.; Manetti, R.; Dolei, A. Expression and activation by Epstein Barr virus of human endogenous retroviruses-W in blood cells and astrocytes: Inference for multiple sclerosis. PLoS ONE 2012, 7, e44991, doi:10.1371/journal.pone.0044991.
- Li,W.; Lee, M.-H.; Henderson, L.; Tyagi, R.; Bachani, M.; Steiner, J.; Campanac, E.; Hoffman, D.A.; Geldern, G.V.; Johnson, K.; et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci. Transl. Med. 2015, 7, 307ra153
- Karlsson, H.; Bachmann, S.; Schröder, J.; McArthur, J.; Torrey, E.F.; Yolken, R.H. Retroviral RNA identified in the cerebrospinal fluids and brains of individuals with schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4634–4639, doi:10.1073/pnas.061021998.
- Slokar, G.; Hasler, G. Human endogenous retroviruses as pathogenic factors in the development of schizophrenia. Front. Psychiatry 2015, 6, 183, doi:10.3389/fpsyt.2015.00183.
- Johansson, E.M.; Bouchet, D.; Tamouza, R.; Ellul, P.; Morr, A.S.; Avignone, E.; Germi, R.; Leboyer, M.; Perron, H.; Groc, L. Human endogenous retroviral protein triggers deficit in glutamate synapse maturation and behaviors associated with psychosis. Sci. Adv. 2020 6, eabc0708, doi:10.1126/sciadv.abc0708
- Kury, P.; Nath, A.; Creange, A.; Dolei, A.; Marche, P.; Gold, J.; Giovannoni, G.; Hartung, H.P.; Perron, H. Human Endog-enous Retroviruses in Neurological Diseases. Trends Mol. Med. 2018, 24, 379–394, doi:10.1016/j.molmed.2018.02.007.
- Blond, J.L.; Besème, F.; Duret, L.; Bouton, O.; Bedin, F.; Perron, H.; Mandrand, B.; Mallet, F. Molecular characterization and placental expression of HERV-W, a new human endogenous retrovirus family. J. Virol. 1999, 73, 1175–1185, doi:10.1128/JVI.73.2.1175-1185.1999.
- Pavlícek, A.; Paces, J.; Elleder, D.; Hejnar, J. Processed pseudogenes of human endogenous retroviruses generated by LINEs: Their integration, stability, and distribution. Genome Res. 2002, 12, 391–399, doi:10.1101/gr.216902
- Mameli, G.; Astone, V.; Arru, G.; Marconi, S.; Lovato, L.; Serra, C.; Sotgiu, S.; Bonetti, B.; Dolei, A. Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not Human herpesvirus 6. J. Gen. Virol. 2007, 88, 264–274.
- Kim, H.-S.; Kim, D.-S.; Huh, J.-W.; Ahn, K.; Yi, J.-M.; Lee, J.-R.; Hirai, H. Molecular characterization of the HERV-W env gene in humans and primates: Expression, FISH, phylogeny, and evolution. Mol. Cells 2008, 26, 53–60.
- Nowak, J.; Januszkiewicz, D.; Pernak, M.; Liwe´ n, I.; Zawada, M.; Rembowska, J.; Nowicka, K.; Lewandowski, K.; Hertmanoswka, H.; Wender, M. Multiple sclerosis-associated virus-related pol sequences found both in multiple sclerosis and healthy donors are more frequently expressed in multiple sclerosis patients. J. Neurovirol. 2003, 9, 112–117.
- Komurian-Pradel, F.; Paranhos-Baccala, G.; Bedin, F.; Ounanian-Paraz, A.; Sodoyer, M.; Ott, C.; Rajoharison, A.; Garcia, E.; Mallet, F.; Mandrand, B.; et al. Molecular cloning and characterization of MSRV-related sequences associated with retrovi-rus-like particles. Virology 1999, 260, 1–9, doi:10.1006/viro.1999.9792
- Roebke, C.; Wahl, S.; Laufer, G.; Stadelmann, C.; Sauter, M.; Mueller-Lantzsch, N.; Mayer, J.; Ruprecht, K. An N-terminally truncated envelope protein encoded by a human endogenous retrovirus W locus on chromosome Xq22.3. Retrovirology 2010, 7, 69, doi:10.1186/1742-4690-7-69.
- Ruprecht, K.; Mayer, J. On the origin of a pathogenic HERV-W envelope protein present in multiple sclerosis lesions. Proc. Natl. Acad. Sci. USA 2019, 116, 19791–19792, doi:10.1073/pnas.1911703116.
- Marchi, E.; Kanapin, A.; Magiorkinis, G.; Belshaw, R. Unfixed endogenous retroviral insertions in the human population. J. Virol. 2014, 88, 9529–9537, doi:10.1128/JVI.00919-14.
- Serra, C.; Mameli, G.; Arru, G.; Sotgiu, S.; Rosati, G.; Dolei, A. In vitro modulation of the multiple sclerosis (MS)-associated retrovirus by cytokines: Implications for MS pathogenesis. J. Neurovirol. 2003, 9, 637–643, doi:10.1080/13550280390246462.
- Charvet, B.; Reynaud, J.M.; Gourru-Lesimple, G.; Perron, H.; Marche, P.N.; Horvat, B. Induction of Proinflammatory Mul-tiple Sclerosis-Associated Retrovirus Envelope Protein by Human Herpesvirus-6A and CD46 Receptor Engagement. Front. Immunol. 2018, 9, 2803, doi:10.3389/fimmu.2018.02803.
- Charvet, B.; Pierquin, J.; Brunel, J.; Gorter, R.; Quétard, C.; Horvat, B.; Amor, S.; Portoukalian, J.; Perron, H. Human En-dogenous Retrovirus Type W Envelope from Multiple Sclerosis Demyelinating Lesions Shows Unique Solubility and Anti-genic Characteristics. Virol. Sinica. 2021, doi:10.1007/s12250-021-00372-0.
- Wang, X.; Liu, Z.; Wang, P.; Li, S.; Zeng, J.; Tu, X.; Yan, Q.; Xiao, Z.; Pan, M.; Zhu, F. Syncytin-1, an endogenous retroviral protein, triggers the activation of CRP via TLR3 signal cascade in glial cells. Brain Behav. Immun. 2018, 67, 324–334, doi:10.1016/j.bbi.2017.09.009.
- Daskalakis, M.; Brocks, D.; Sheng, Y.-H.; Islam, M.S.; Ressnerova, A.; Assenov, Y.; Milde, T.; Oehme, I.; Witt, O.; Goyal, A.; et al. Reactivation of endogenous retroviral elements via treatment with DNMT- and HDAC-inhibitors. Cell Cycle 2018, 17, 811–822, doi:10.1080/15384101.2018.1442623.
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part I: The role of infection. Ann. Neurol. 2007, 61, 288–299, doi:10.1002/ana.21117.
- Perron, H.; Lang, A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin. Rev. Allergy Immunol. 2010, 39, 51–61, doi:10.1007/s12016-009-8170-x.
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Multiple sclerosis: Autoimmunity and viruses. Curr. Opin. Rheumatol. 2013, 25, 496–501, doi:10.1097/BOR.0b013e328362004d
- Limosin, F.; Rouillon, F.; Payan, C.; Cohen, J-M.; Strub, N. Prenatal exposure to influenza as a risk factor for adult schizo-phrenia: Influenza and schizophrenia. Acta Psychiatr. Scand. 2003, 107, 331–335, doi:10.1034/j.1600-0447.2003.00052.x.
- Shi, L.; Fatemi, S.H.; Sidwell, R.W.; Patterson, P.H. Maternal influenza infection causes marked behavioral and pharmaco-logical changes in the offspring. J. Neurosci. 2003, 23, 297–302, doi:10.1523/JNEUROSCI.23-01-00297.2003.
- Ebert, T.; Kotler, M. Prenatal exposure to influenza and the risk of subsequent development of schizophrenia. Isr. Med. Assoc. J. 2005, 7, 35–38, doi:10.1034/j.1600-0447.2003.00052.x.
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280, doi:10.1176/appi.ajp.2009.09030361.
- Nellåker, C.; Yao, Y.; Jones-Brando, L.; Mallet, F.; Yolken, RH.; Karlsson, H. Transactivation of elements in the human endogenous retrovirus W family by viral infection. Retrovirology 2006, 3, 44, doi:10.1186/1742-4690-3-44.
- Li, F.; Nellåker, C.; Sabunciyan, S.; Yolken, R.H.; Jones-Brando, L.; Johansson, A-S.; et al. Transcriptional derepression of the ERVWE1 locus following influenza A virus infection. J. Virol. 2014, 88, 4328–4337, doi:10.1128/JVI.03628-13.
- Schmidt, N.; Domingues, P.; Golebiowski, F.; Patzina, C.; Tatham, M.H.; Hay, R.T.; Hale, B.G. An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proc. Natl. Acad. Sci. USA 2019, 16, 17399–17408, doi:10.1073/pnas.1907031116.
- Bhende, P.M.; Seaman, W.T.; Delecluse, H-J.; Kenney, S.C. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat. Genet. 2004;36(10):1099–104, doi:10.1038/ng1424.
- Moore, F.G.A.; Wolfson, C. Human herpes virus 6 and multiple sclerosis: Human herpes virus 6 and multiple sclerosis. Acta Neurol. Scand. 2002, 106, 63–83, doi:10.1034/j.1600-0404.2002.01251.x.
- Manghera, M.; Ferguson-Parry, J.; Lin, R.; Douville, R.N. NF-κB and IRF1 induce endogenous retrovirus K expression via interferon-stimulated response elements in its 5′ long terminal repeat. J. Virol. 2016;90, 9338–9349, doi:10.1128/JVI.01503-16.
- Brown, A.S. The environment and susceptibility to schizophrenia. Prog. Neurobiol. 2011, 93, 23–58, doi:10.1016/j.pneurobio.2010.09.003.
- Waubant, E.; Lucas, R.; Mowry, E.; Graves, J.; Olsson, T.; Alfredsson, L.; Langer-Gould, A. Environmental and genetic risk factors for MS: An integrated review. Ann. Clin. Transl. Neurol. 2019, 6, 1905–1922, doi:10.1002/acn3.50862.
- Garcia-Montojo, M.; Dominguez-Mozo, M.; Arias-Leal, A.; Garcia-Martinez, Á.; De las Heras, V.; Casanova, I.; Faucard, R.; Gehin, N.; Madeira, A.; Arroyo, R.; et al. The DNA copy number of human endogenous retrovirus-W (MSRV-type) is in-creased in multiple sclerosis patients and is influenced by gender and disease severity. PLoS ONE 2013, 8, e53623, doi:10.1371/journal.pone.0053623.
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106, doi:10.1111/j.1476-5381.2011.01302.x.
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsy-chiatr. Dis. Treat. 2017, 13, 1661–1667, doi:10.2147/NDT.S140634.
- Goldmann, T.; Prinz, M. Role of microglia in CNS autoimmunity. Clin. Dev. Immunol. 2013, 2013, 209093, doi:10.1155/2013/208093
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11,374, doi:10.3389/fimmu.2020.00374.
- Monoghan, K.L.; Zheng, W.; Hu, G.; Wan, E.C.K. Monocytes and Monocyte-Derived Antigen-Presenting Cells Have Distinct Gene Signatures in Experimental Model of Multiple Sclerosis. Front. Immunol. 2019, 10, 2779, doi:10.3389/fimmu.2019.02779
- Engelhardt, B.; Ransohoff, R.M. apture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends. Immunol. 2012, 33, 579–589, doi:10.1016/j.it.2012.07.004.
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635, doi:10.1038/nri3265.
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783, doi:10.1126/science.aag2590.
- Ludewig, P.; Gallizioli, M.; Urra, X.; Behr, S.; Brait, V.H.; Gelderblom, M.; Magnus, T.; Planas, A.M. Dendritic cells in brain diseases. Biochim. Biophys. Acta 2016, 1862, 352–367, doi:10.1016/j.bbadis.2015.11.003.
- McFarland, H.F.; Martin, R. Multiple sclerosis: A complicated picture of autoimmunity. Nat. Immunol. 2007, 8, 913–919, doi:10.1038/ni1507.
- Martino, G.; Adorini, L.; Rieckmann, P.; Hillert, J.; Kallmann, B.; Comi, G.; Filippi, M. Inflammation in multiple sclerosis: The good, the bad, and the complex. Lancet Neurol. 2002, 1, 499–509, doi:10.1016/s1474-4422(02)00223-5.
- Hemmer, B.; Nessler, S.; Zhou, D.; Kieseier, B.; Hartung, H.-P. Immunopathogenesis and immunotherapy of multiple sclerosis. Nat. Clin. Pract. Neurol. 2006, 2, 201–211, doi:10.1038/ncpneuro0154.
- Popescu, B.F.G.; Lucchinetti, C.F. Pathology of demyelinating diseases. Annu. Rev. Pathol. 2012, 7, 185–217, doi:10.1146/annurev-pathol-011811-132443.
- Duperray, A.; Barbe, D.; Raguenez, G.; Weksler, B.B.; Romero, I.A.; Couraud, P-O.; Perron, H.; Marche, P.N. Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int. Immunol. 2015, 27, 545–553, doi:10.1093/intimm/dxv025.
- Kremer, D.; Schichel, T.; Förster, M.; Tzekova, N.; Bernard, C.; van der Valk, P.; Horssen, J.V.; Hartung, H-P.; Perron, H.; Küry, P. Human endogenous retrovirus type W envelope protein inhibits oligodendroglial precursor cell differentiation: Retroviruses and Myelin Repair. Ann. Neurol. 2013, 74, 721–732, doi:10.1002/ana.23970.
- Kremer, D.; Gruchot, J.; Weyers, V.; Oldemeier, L.; Göttle, P.; Healy, L.; Jang, J.H.; Xu, Y.K.T.; Volsko, C.; Dutta, R.; et al. pHERV-W envelope protein fuels microglial cell-dependent damage of myelinated axons in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2019, 116, 15216–15225.
- Göttle, P.; Förster, M.; Gruchot, J.; Kremer, D.; Hartung, H.P.; Perron, H.; Küry, P. Rescuing the negative impact of human endogenous retrovirus envelope protein on oligodendroglial differentiation and myelination. Glia 2019, 67, 160–170, doi:10.1002/glia.23535.
- Perron, H.; Dougier-Reynaud, H-L.; Lomparski, C.; Popa, I.; Firouzi, R.; Bertrand, J-B.; Marusic, S.; Portoukalian, J.; Jouvin-Marche, E.; Villiers, C.L.; et al. Human endogenous retrovirus protein activates innate immunity and promotes ex-perimental allergic encephalomyelitis in mice. PLoS ONE 2013, 8, e80128, doi:10.1371/journal.pone.0080128.
- Perron, H.; Germi, R.; Bernard, C.; García-Montojo, M.; Deluen, C.; Farinelli, L.; Faucard, R.; Veas, F.; Stefas, I.; Fabriek, B.O; et al. Human endogenous retrovirus type W envelope expression in blood and brain cells provides new insights into multiple sclerosis disease. Mult. Scler. 2012, 18, 1721–1736, doi:10.1177/1352458512441381.
- Perron, H.; Lazarini, F.; Ruprecht, K.; Péchoux-Longin, C.; Seilhean, D.; Sazdovitch, V.; Créange, A.; Battail-Poirot, N.; Sibaï, G.; Santoro, L.; et al. Human endogenous retrovirus (HERV)-W ENV and GAG proteins: Physiological expression in human brain and pathophysiological modulation in multiple sclerosis lesions. J. Neurovirol. 2005, 11, 23–33, doi:10.1080/13550280590901741.
- Brudek, T.; Christensen, T.; Aagaard, L.; Petersen, T.; Hansen, H.J.; Møller-Larsen, A. B cells and monocytes from patients with active multiple sclerosis exhibit increased surface expression of both HERV-H Env and HERV-W Env, accompanied by increased seroreactivity. Retrovirology 2009, 6, 104.
- Kremer, D.; Schichel, T.; Förster, M.; Tzekova, N.; Bernard, C.; van der Valk, P.; Horssen, J.V.; Hartung, H.-P.; Perron, H.; Küry, P. Human endogenous retrovirus type W envelope protein inhibits oligodendroglial precursor cell differentiation: Retroviruses and Myelin Repair. Ann. Neurol. 2013, 74, 721–732.
- Garson, J.A.; Tuke, P.W.; Giraud, P.; Paranhos-Baccala, G.; Perron, H. Detection of virion-associated MSRV-RNA in serum of patients with multiple sclerosis. Lancet 1998, 351, 33, doi:10.1016/s0140-6736(98)24001-3.
- Dolei, A.; Serra, C.; Mameli, G.; Pugliatti, M.; Sechi, G.; Cirotto, M.C.; Rosati, G.; Sotgiu, S. Multiple sclerosis-associated retrovirus (MSRV) in Sardinian MS patients. Neurology 2002, 58, 471–473, doi:10.1212/wnl.58.3.471.
- Sotgiu, S.; Arru, G.; Mameli, G.; Serra, C.; Pugliatti, M.; Rosati, G.; Dolei, A. Multiple sclerosis-associated retrovirus in early multiple sclerosis: A six-year follow-up of a Sardinian cohort. Mult. Scler. 2006, 12, 698–703, doi:10.1177/1352458506070773.
- Mameli, G.; Serra, C.; Astone, V.; Castellazzi, M.; Poddighe, L.; Fainardi, E.; Neri, W.; Granieri, W.; Dolei, A. Inhibition of multiple-sclerosis-associated retrovirus as biomarker of interferon therapy. J. Neurovirol. 2008;14, 73–77, doi:10.1080/13550280701801107.
- Sotgiu, S.; Pugliatti, M.; Sanna, A.; Sotgiu, A.; Castiglia, P.; Solinas, G.; Dolei, A.; Serra, C.; Bonetti, B.; Rosati, G. Multiple sclerosis complexity in selected populations: The challenge of Sardinia, insular Italy. Eur. J. Neurol. 2002, 9, 329–341, doi:10.1046/j.1468-1331.2002.00412.x
- Sotgiu, S.; Mameli, G.; Serra, C.; Zarbo, I.R.; Arru, G.; Dolei, A. Multiple sclerosis-associated retrovirus and progressive disability of multiple sclerosis. Mult. Scler. 2010, 16, 1248–1251, doi:10.1177/1352458510376956.
- Gerondakis, S.; Grumont, R.J.; Banerjee, A. Regulating B‐cell activation and survival in response to TLR signals. Immunol. Cell Biol. 2007, 85, 471–475, doi:10.1038/sj.icb.7100097.
- Firouzi, R.; Rolland, A.; Michel, M.; Jouvin-Marche, E.; Hauw, J.J.; Malcus-Vocanson, C.; Lazarini, F.; Gebuhrer, L.; Seigneurin, J.M.; Touraine, J.L.; et al. Multiple sclerosis-associated retrovirus particles cause T lymphocyte-dependent death with brain hemorrhage in humanized SCID mice model. J. Neurovirol. 2003, 9, 79–93.
- Kaisho, T.; Akira, S. Toll-like receptor function and signaling. J. Allergy Clin. Immunol. 2006, 117, 979–987, doi:10.1016/j.jaci.2006.02.023.
- Bsibsi, M.; Ravid, R.; Gveric, D.; van Noort, J.M. Broad expression of Toll-like receptors in the human central nervous system. J. Neuropathol. Exp. Neurol. 2002, 61, 1013–1021, doi:10.1093/jnen/61.11.1013.
- Bowman, C.C.; Rasley, A.; Tranguch, S.L.; Marriott, I. Cultured astrocytes express toll-like receptors for bacterial products. Glia 2003, 43, 281–291, doi:10.1002/glia.10256.
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924, doi:10.4049/jimmunol.173.6.3916.
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216, doi:10.1146/annurev.immunol.20.083001.084359.
- Akira, S.; Hemmi, H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol. Lett. 2003, 85, 85–95, doi:10.1016/s0165-2478(02)00228-6.
- Planas, R.; Metz, I.; Martin, R, Sospedra, M. Detailed Characterization of T Cell Receptor Repertoires in Multiple Sclerosis Brain Lesions. Front. Immunol. 2018, 9, 509, doi:10.3389/fimmu.2018.00509.
- Sospedra, M.; Martin, R. Immunology of multiple sclerosis. Semin. Neurol. 2016, 36, 115–127.
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.A.; Volpe, J.K.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519.
- Franklin, R.J.M.; Ffrench-Constant, C. Regenerating CNS myelin-from mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017, 18, 753–769, doi:10.1038/nrn.2017.136.
- Touil, T.; Deloire-Grassin, M.S.; Vital, C.; Petry, K.G.; Brochet, B. In vivo damage of CNS myelin and axons induced by peroxynitrite. Neuroreport 2001, 12, 3637–3644, doi:10.1097/00001756-200111160-00052.
- Karamita, M.; Barnum, C.; Möbius, W.; Tansey, M.G.; Szymkowski, D.E.; Lassmann, H.; Probert, L. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight. 2017, 2, doi:10.1172/jci.insight.87455
- Madeira, A.; Burgelin, I.; Perron, H.; Curtin, F.; Lang, A.B.; Faucard, R. MSRV envelope protein is a potent, endogenous and pathogenic agonist of human toll-like receptor 4: Relevance of GNbAC1 in multiple sclerosis treatment. J. Neuroimmunol. 2016, 291, 29–38, doi:10.1016/j.jneuroim.2015.12.006.
- Faucard, R.; Madeira, A.; Gehin, N.; Authier, F-J.; Panaite, P-A.; Lesage, C.; Burgelin, I.; Bertel, M.; Bernard, C.; Curtin, F.; et al. Human endogenous retrovirus and neuroinflammation in chronic inflammatory demyelinating polyradiculoneuropathy. EBioMedicine 2016, 6, 190–198, doi:10.1016/j.ebiom.2016.03.001.
- Christensen, T. HERVs in neuropathogenesis. J. Neuroimmune Pharmacol. 2010, 5, 326–335, doi:10.1007/s11481-010-9214-y.
- Leboyer, M.; Tamouza, R.; Charron, D.; Faucard, R.; Perron, H. Human endogenous retrovirus type W (HERV-W) in schizophrenia: A new avenue of research at the gene-environment interface. World J. Biol. Psychiatry 2011, 14, 80–90, doi:10.3109/15622975.2010.601760.
- Howes, O.D.; McCutcheon, R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: A reconceptual-ization. Transl. Psychiatry 2017, 7, e1024–e1024, doi:10.1038/tp.2016.278.
- Müller, N.; Wagner, J.K.; Krause, D.; Weidinger, E.; Wildenauer, A.; Obermeier, M.; Dehning, S.; Gruber, R.; Shwarz, M.J. Impaired monocyte activation in schizophrenia. Psychiatry Res. 2012, 198, 341–346, doi:10.1016/j.psychres.2011.12.049.
- Potvin, S.; Stip, E.; Sepehry, A.A.; Gendron, A.; Bah, R.; Kouassi, E. Inflammatory cytokine alterations in schizophrenia: A systematic quantitative review. Biol. Psychiatry 2008, 63, 801–808, doi:10.1016/j.biopsych.2007.09.024.
- Misiak, B.; Stańczykiewicz, B.; Kotowicz, K.; Rybakowski, J.K.; Samochowiec, J.; Frydecka, D. Cytokines and C-reactive protein alterations with respect to cognitive impairment in schizophrenia and bipolar disorder: A systematic review. Schiz-ophr. Res. 2018, 192, 16–29, doi:10.1016/j.schres.2017.04.015.
- Dickerson, F.; Stallings, C.; Origoni, A.; Boronow, J.; Yolken, R. C-reactive protein is associated with the severity of cognitive impairment but not of psychiatric symptoms in individuals with schizophrenia. Schizophr. Res. 2007, 93, 261–265, doi:10.1016/j.schres.2007.03.022.
- Melbourne, J.K.; Chase, K.A.; Feiner, B.; Rosen, C.; Sharma, R.P. Long non-coding and endogenous retroviral RNA levels are associated with proinflammatory cytokine mRNA expression in peripheral blood cells: Implications for schizophrenia. Psy-chiatry Res. 2018, 262, 465–468, doi:10.1016/j.psychres.2017.09.025.
- Karlsson, H.; Schröder, J.; Bachmann, S.; Bottmer, C.; Yolken, R.H. HERV-W-related RNA detected in plasma from individuals with recent-onset schizophrenia or schizoaffective disorder. Mol. Psychiatry 2004, 9, 12–13.
- Perron, H.; Mekaoui, L.; Bernard, C.; Veas, F.; Stefas, I.; Leboyer, M. Endogenous retrovirus typeWGAG and envelope protein antigenemia in serum of szhizophrenia patients. Biol. Psychiatry 2008, 64, 1019–1023
- Huang, W.; Li, S.; Hu, Y.; Yu, H.; Luo, F.; Zhang, Q.; Zhu, F. Implication of the env gene of the human endogenous retrovirus W family in the expression of BDNF and DRD3 and development of recent-onset schizophrenia. Schizophr. Bull. 2011, 37, 988–1000.
- Deb-Rinker, P.; Klempan, T.A.; O’Reilly, R.L.; Torrey, E.F.; Singh, S.M. Molecular characterization of a MSRV-like sequence identified by RDA from monozygotic twin pairs discordant for schizophrenia. Genomics 1999, 61, 133–144, doi:10.1006/geno.1999.5946.
- Yao, Y.; Schröder, J.; Nellåker, C.; Bottmer, C.; Bachmann, S.; Yolken, R.H.; Karlsson, H. Elevated levels of human endog-enous retrovirus-W transcripts in blood cells from patients with first episode schizophrenia. Genes Brain Behav. 2008, 7, 103–112, doi:10.1111/j.1601-183X.2007.00334.x.
- Yolken, R.H.; Karlsson, H.; Yee, F.; Johnston-Wilson, N.L.; Torrey, E.F. Endogenous retroviruses and schizophrenia. Brain Res. Rev. 2000, 31, 193–199, doi:10.1016/s0165-0173(99)00037-5.
- Gourion, D.; Goldberger, C.; Leroy, S.; Bourdel, M-C.; Olié, J-P.; Krebs, M-O. Age at onset of schizophrenia: Interaction between brain-derived neurotrophic factor and dopamine D3 receptor gene variants. Neuroreport 2005, 16, 1407–1410, doi:10.1097/01.wnr.0000175245.58708.6b.