Nonsense-mediated messenger RNA (mRNA) decay (NMD) is a surveillance pathway used by cells to control the quality mRNAs and to fine-tune transcript abundance. NMD plays an important role in cell cycle regulation, cell viability, DNA damage response, while also serving as a barrier to virus infection. Disturbance of this control mechanism caused by genetic mutations or dys-regulation of the NMD pathway can lead to pathologies, including neurological disorders, immune diseases and cancers. The role of NMD in cancer development is complex, acting as both a promoter and a barrier to tumour progression. Cancer cells can exploit NMD for the downregulation of key tumour suppressor genes, or tumours adjust NMD activity to adapt to an aggressive immune microenvironment.
- Nonsense-mediated mRNA decay
- Premature termination codon
- Cancer
- Neoantigens
- NMD inhibition
1. The Nonsense-Mediated mRNA Decay (NMD) Pathway and Machinery
1.1. The NMD Machinery
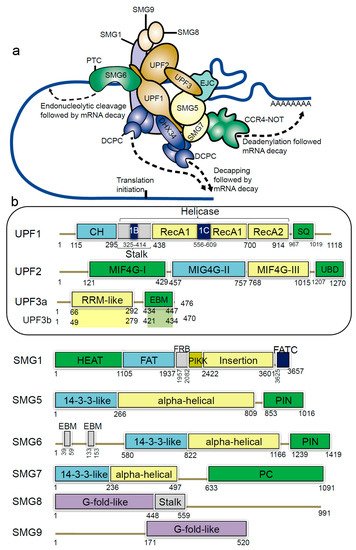
1.2. Structural Insights of NMD Components at a Glance
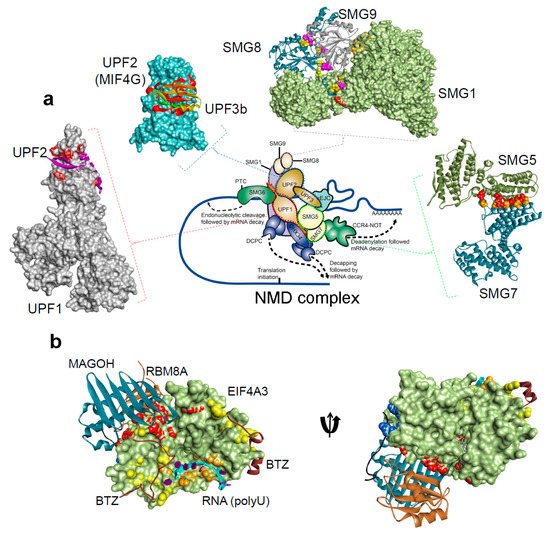
| Protein Interacting Partners | PDB ID. | Resolution | Method | References |
|---|---|---|---|---|
| UPF1-UPF2 | 2wjv | 2.85 Å | X-Ray diffraction | [34] |
| UPF2-UPF3b | 1uw4 | 1.95 Å | X-Ray diffraction | [36] |
| SMG5-SMG7 | 3zhe | 3 Å | X-Ray diffraction | [48] |
| SMG8-SMG9 | 5nkk | 2.64 Å | X-Ray diffraction | [42] |
| SMG1–SMG8–SMG9 | 6syt | 3.45 Å | Electron microscopy | [43] |
| Mago-Y14-eIF4AIII-Barentsz-UPF3b | 2xb2 | 3.4 Å | X-Ray diffraction | [44] |
1.3. NMD Target Selection, more than just Coding Transcripts
2. NMD as a Crucial Regulator of the Transcriptome
2.1. NMD in the Maintenance and Homeostasis of the Cell
2.2. NMD Factors are Essential in Embryonic Development
2.3. Cellular Responses to Stress through NMD Regulation
2.4. NMD as a Regulator of the Immune Response and Viral Replication
3. Nonsense-Mediated mRNA Decay and Genetic Disease
4. A Dual Role for NMD in Cancer
4.1. NMD as a Protective Agent in Cancer
4.2. NMD Implied in Cancer Aggressiveness and Progression
4.3. Role of NMD in the Tumour Microenvironment
References
- Nickless, A.; Bailis, J.M.; You, Z. Control of gene expression through the nonsense-mediated RNA decay pathway. Cell Biosci. 2017, 7, 26.
- Popp, M.W.; Maquat, L.E. Nonsense-mediated mRNA Decay and Cancer. Curr. Opin. Genet. Dev. 2018, 48, 44–50.
- He, F.; Li, X.; Spatrick, P.; Casillo, R.; Dong, S.; Jacobson, A. Genome-Wide Analysis of mRNAs Regulated by the Nonsense-Mediated and 5′ to 3′ mRNA Decay Pathways in Yeast. Mol. Cell 2003, 12, 1439–1452.
- Celik, A.; Kervestin, S.; Jacobson, A. NMD: At the crossroads between translation termination and ribosome recycling. Biochimie 2015, 114, 2–9.
- Amrani, N.; Ganesan, R.; Kervestin, S.; Mangus, D.A.; Ghosh, S.; Jacobson, A. A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 2004, 7013, 112–118.
- Gatfield, D.; Unterholzner, L.; Ciccarelli, F.D.; Bork, P.; Izaurralde, E. Nonsense-mediated mRNA decay in Drosophila: At the intersection of the yeast and mammalian pathways. EMBO J. 2003, 22, 3960–3970.
- Wang, Z.; Ballut, L.; Barbosa, I.; Le Hir, H. Exon Junction Complexes can have distinct functional flavours to regulate specific splicing events. Sci. Rep. 2018, 8, 9509.
- Buhler, M.; Steiner, S.; Mohn, F.; Paillusson, A.; Muhlemann, O. EJC-independent degradation of nonsense immunoglobulin-μ mRNA depends on 3′ UTR length. Nat. Struct. Mol. Biol. 2006, 13, 462–464.
- Pulak, R.; Anderson, P. mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev. 1993, 7, 1885–1897.
- Leeds, P.; Wood, J.M.; Lee, B.S.; Culbertson, M.R. Gene products that promote mRNA turnover in Saccharomyces cerevisiae. Mol. Cell. Biol. 1992, 12, 2165–2177.
- Muhlemann, O.; Eberle, A.B.; Stalder, L.; Zamudio Orozco, R. Recognition and elimination of nonsense mRNA. Biochim. Biophys. Acta 2008, 1779, 538–549.
- Boehm, V.; Gehring, N.H. Exon Junction Complexes: Supervising the Gene Expression Assembly Line. Trends Genet. 2016, 11, 724–735.
- Bono, F.; Ebert, J.; Lorentzen, E.; Conti, E. The Crystal Structure of the Exon Junction Complex Reveals How It Maintains a Stable Grip on mRNA. Cell 2006, 126, 713–725.
- Le Hir, H.; Izaurralde, E.; Maquat, L.E.; Moore, M.J. The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000, 24, 6860–6869.
- Gardner, L.B. Nonsense mediated RNA decay regulation by cellular stress; implications for tumorigenesis NIH Public Access. Mol. Cancer Res. 2010, 8, 295–308.
- Ishigaki, Y.; Li, X.; Serin, G.; Maquat, L.E. Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell 2001, 106, 607–617.
- Dostie, J.; Dreyfuss, G. Translation is required to remove Y14 from mRNAs in the cytoplasm. Curr. Biol. 2002, 13, 1060–1067.
- Lykke-Andersen, J.; Shu, M.D.; Steitz, J.A. Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 2000, 103, 1121–1131.
- Gehring, N.H.; Neu-Yilik, G.; Schell, T.; Hentze, M.W.; Kulozik, A.E. Y14 and hUpf3b form an NMD-activating complex. Mol. Cell 2003, 11, 939–949.
- Cheng, Z.; Saito, K.; Pisarev, A.V.; Wada, M.; Pisareva, V.P.; Pestova, T.V.; Gajda, M.; Round, A.; Kong, C.; Lim, M. Structural insights into eRF3 and stop codon recognition by eRF1. Genes Dev. 2009, 23, 1106–1118.
- Lykke-Andersen, S.; Jensen, T.H. Nonsense-mediated mRNA decay: An intricate machinery that shapes transcriptomes. Nat. Publ. Group 2015, 16, 665–677.
- Karousis, E.D.; Nasif, S.; Mühlemann, O. Nonsense-mediated mRNA decay: Novel mechanistic insights and biological impact. RNA 2016, 7, 661–682.
- Kalathiya, U.; Padariya, M.; Pawlicka, K.; Verma, C.S.; Houston, D.; Hupp, T.R.; Alfaro, J.A. Insights into the Effects of Cancer Associated Mutations at the UPF2 and ATP-Binding Sites of NMD Master Regulator: UPF1. Int. J. Mol. Sci. 2019, 20, 5644.
- Kashima, I.; Yamashita, A.; Izumi, N.; Kataoka, N.; Morishita, R.; Hoshino, S.; Ohno, M.; Dreyfuss, G.; Ohno, S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev. 2006, 20, 355–367.
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259.
- Chan, W.K.; Bhalla, A.D.; Le Hir, H.; Nguyen, L.S.; Huang, L.; Gécz, J.; Wilkinson, M.F. A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat. Struct. Mol. Biol. 2009, 16, 747–753.
- Isken, O.; Kim, Y.K.; Hosoda, N.; Mayeur, G.L.; Hershey, J.W.B.; Maquat, L.E. Upf1 Phosphorylation Triggers Translational Repression during Nonsense-Mediated mRNA Decay. Cell 2008, 133, 314–327.
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467.
- Muhlrad, D.; Parker, R. Premature translational termination triggers mRNA decapping. Nature 1994, 370, 578–581.
- Huntzinger, E.; Kashima, I.; Fauser, M.; Saulière, J.; Izaurralde, E. SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 2008, 14, 2609–2617.
- Melero, R.; Hug, N.; López-Perrote, A.; Yamashita, A.; Cáceres, J.F.; Llorca, O. The RNA helicase DHX34 functions as a scaffold for SMG1-mediated UPF1 phosphorylation. Nat. Commun. 2016, 7, 10585.
- Hug, N.; Caceres, J.F. The RNA helicase DHX34 activates NMD by promoting a transition from the surveillance to the decay-inducing complex. Cell Rep. 2014, 8, 1845–1856.
- Lejeune, F.; Li, X.; Maquat, L.E. Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol. Cell 2003, 12, 675–687.
- Clerici, M.; Mourão, A.; Gutsche, I. Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J. 2009, 28, 2293–2306.
- Chakrabarti, S.; Jayachandran, U.; Bonneau, F.; Fiorini, F.; Basquin, C.; Domcke, S.; Le Hir, H.; Conti, E. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell 2011, 41, 693–703.
- Kadlec, J.; Izaurralde, E.; Cusack, S. The structural basis for the interaction between nonsense-mediated mRNA decay factors UPF2 and UPF3. Nat. Struct. Mol. Biol. 2004, 11, 330–337.
- Clerici, M.; Deniaud, A.; Boehm, V.; Gehring, N.H.; Schaffitzel, C.; Cusack, S. Structural and functional analysis of the three MIF4G domains of nonsense-mediated decay factor UPF2. Nucleic Acids Res. 2013, 42, 2673–2686.
- Gupta, P.; Li, Y. Upf proteins: Highly conserved factors involved in nonsense mRNA mediated decay. Mol. Biol. Rep. 2018, 45, 39–55.
- Bhuvanagiri, M.; Lewis, J.; Putzker, K.; Becker, J.P.; Leicht, S.; Krijgsveld, J.; Batra, R.; Turnwald, B.; Jovanovic, B.; Hauer, C. 5-azacytidine inhibits nonsense-mediated decay in a MYC-dependent fashion. EMBO Mol. Med. 2014, 6, 1593–1609.
- Wang, D.; Wengrod, J.; Gardner, L.B. Overexpression of the c-myc oncogene inhibits nonsense-mediated RNA decay in B lymphocytes. J. Biol. Chem. 2011, 286, 40038–40043.
- Loh, B.; Jonas, S.; Izaurralde, E. The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev. 2013, 27, 2125–2138.
- Li, L.; Lingaraju, M.; Basquin, C.; Basquin, J.; Conti, E. Structure of a SMG8–SMG9 complex identifies a G-domain heterodimer in the NMD effector proteins. RNA 2017, 23, 1028–1034.
- Gat, Y.; Schuller, J.M.; Lingaraju, M.; Weyher, E.; Bonneau, F.; Strauss, M.; Murray, P.J.; Conti, E. InsP6 binding to PIKK kinases revealed by the cryo-EM structure of an SMG1-SMG8-SMG9 complex. Nat. Struct. Mol. Biol. 2019, 26, 1089–1093.
- Buchwald, G.; Ebert, J.; Basquin, C.; Sauliere, J.; Jayachandran, U.; Bono, F.; Conti, E. Insights into the Recruitment of the NMD Machinery from the Crystal Structure of a Core EJC-UPF3b Complex. Proc. Natl. Acad. Sci. USA 2010, 107, 10050–10055.
- Frischmeyer, P.A.; Dietz, H.C. Nonsense-mediated mRNA decay in health and disease. Hum. Mol. Genet. 1999, 8, 1893–1900.
- Jin, L.; Wang, W.; Fang, G. Targeting protein-protein interaction by small molecules. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 435–456.
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlić, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned website and web services. Nucleic Acids Res. 2011, 39, 392–401.
- Jonas, S.; Weichenrieder, O.; Izaurralde, E. An unusual arrangement of two 14-3-3-like domains in the SMG5-SMG7 heterodimer is required for efficient nonsense-mediated mRNA decay. Genes Dev. 2013, 27, 211–225.
- Page, M.F.; Carr, B.; Anders, K.R.; Grimson, A.; Anderson, P. SMG-2 Is a Phosphorylated Protein Required for mRNA Surveillance in Caenorhabditis elegans and Related to Upf1p of Yeast. Mol. Cell. Biol. 1999, 19, 5943–5951.
- Fukuhara, N.; Ebert, J.; Unterholzner, L.; Lindner, D.; Izaurralde, E.; Conti, E. SMG7 is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol. Cell 2015, 17, 537–547.
- Okada-Katsuhata, Y.; Yamashita, A.; Kutsuzawa, K.; Izumi, N.; Hirahara, F.; Ohno, S. N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res. 2012, 40, 1251–1266.
- Eberle, A.; Lykke-Andersen, S.; Mühlemann, O. SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat. Struct. Mol. Biol. 2009, 16, 49–55.
- Colombo, M.; Karousis, E.D.; Bourquin, J.; Bruggmann, R.; Muhlemann, O. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA 2017, 23, 189–201.
- Smith, J.E.; Alvarez-Dominguez, J.R.; Kline, N.; Huynh, N.J.; Geisler, S.; Hu, W.; Baker, K.E. Translation of small open reading frames within unannotated RNA transcripts in Saccharomyces cerevisiae. Cell Rep. 2014, 7, 1858–1866.
- Lykke-Andersen, S.; Chen, Y.; Ardal, B.R.; Lilje, B.; Waage, J.; Sandelin, A.; Jensen, T.H. Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev. 2014, 28, 2498–2517.
- Smith, J.E.; Baker, K.E. Nonsense-mediated RNA decay--a switch and dial for regulating gene expression. Bioessays 2015, 37, 612–623.
- Hug, N.; Longman, D.; Caceres, J.F. Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 2016, 44, 1483–1495.
- Azzalin, C.M.; Lingner, J. The human RNA surveillance factor UPF1 is required for S phase progression and genome stability. Curr. Biol. 2006, 16, 433–439.
- Nelson, J.O.; Moore, K.A.; Chapin, A.; Hollien, J.; Metzstein, M.M. Degradation of Gadd45 mRNA by nonsense-mediated decay is essential for viability. Elife 2016, 5, e12876.
- Martin, L.; Gardner, L.B. Stress-induced inhibition of nonsense-mediated RNA decay regulates intracellular cystine transport and intracellular glutathione through regulation of the cystine/glutamate exchanger SLC7A11. Oncogene 2015, 34, 4211–4218.
- Gewandter, J.S.; Bambara, R.A.; O’Reilly, M.A. The RNA surveillance protein SMG1 activates p53 in response to DNA double-strand breaks but not exogenously oxidized mRNA. Cell Cycle 2011, 10, 2561–2567.
- Balistreri, G.; Horvath, P.; Schweingruber, C.; Zünd, D.; McInerney, G.; Merits, A.; Helenius, A. The host nonsense-mediated mRNA decay pathway restricts mammalian RNA virus replication. Cell Host Microbe 2014, 16, 403–411.
- Karam, R.; Wengrod, J.; Gardner, L.B.; Wilkinson, M.F. Regulation of nonsense-mediated mRNA decay: Implications for physiology and disease. Biochim. Biophys. Acta Gene Regul. Mech. 2013, 1829, 624–633.
- Fatscher, T.; Boehm, V.; Gehring, N.H. Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell. Mol. Life Sci. 2015, 72, 4523–4544.
- Lou, C.H.; Dumdie, J.; Goetz, A.; Shum, E.Y.; Brafman, D.; Liao, X.; Wilkinson, M.F. Nonsense-mediated RNA decay influences human embryonic stem cell fate. Stem Cell Rep. 2016, 6, 844–857.
- Medghalchi, S.M.; Frischmeyer, P.A.; Mendell, J.T.; Kelly, A.G.; Lawler, A.M.; Dietz, H.C. Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum. Mol. Genet. 2001, 10, 99–105.
- Weischenfeldt, J.; Waage, J.; Tian, G.; Zhao, J.; Damgaard, I.; Jakobsen, J.S. Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol. 2012, 13, R35.
- Li, T.; Shi, Y.; Wang, P.; Guachalla, L.M.; Sun, B.; Joerss, T.; Wang, Z. Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. 2015, 34, 1630–1647.
- Mcilwain, D.R.; Pan, Q.; Reilly, P.T.; Elia, A.J.; Mccracken, S.; Wakeham, A.C.; Mak, T.W. Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc. Natl. Acad. Sci. USA 2010, 107, 12186–12191.
- Wang, D.; Zavadil, J.; Martin, L.; Parisi, F.; Friedman, E.; Levy, D.; Gardner, L.B. Inhibition of Nonsense-Mediated RNA Decay by the Tumor Microenvironment Promotes Tumorigenesis. Mol. Cell. Biol. 2011, 31, 3670–3680.
- Gardner, L.B. Hypoxic Inhibition of Nonsense-Mediated RNA Decay Regulates Gene Expression and the Integrated Stress Response. Mol. Cell. Biol. 2008, 28, 3729–3741.
- Mino, T.; Murakawa, Y.; Fukao, A.; Vandenbon, A.; Wessels, H.H.; Ori, D.; Uehata, T.; Tartey, S.; Akira, S.; Suzuki, Y. Regnase-1 and Roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell 2015, 161, 1058–1073.
- Belew, A.T.; Meskauskas, A.; Musalgaonkar, S.; Advani, V.M.; Sulima, S.O.; Kasprzak, W.K.; Shapiro, B.A.; Dinman, J.D. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 2014, 512, 265–269.
- Quek, B.L.; Beemon, K. Retroviral strategy to stabilize viral RNA. Curr. Opin. Microbiol. 2014, 18, 78–82.
- Mocquet, V.; Neusiedler, J.; Rende, F.; Cluet, D.; Robin, J.P.; Terme, J.M.; Duc Dodon, M.; Wittmann, J.; Morris, C.; Le Hir, H. The human T-lymphotropic virus type 1 tax protein inhibits nonsense-mediated mRNA decay by interacting with INT6/EIF3E and UPF1. J. Virol. 2012, 86, 7530–7543.
- Garcia, D.; Garcia, S.; Voinnet, O. Nonsense-mediated decay serves as a general viral restriction mechanism in plants. Cell Host Microbe 2014, 16, 391–402.
- Wada, M.; Lokugamage, K.G.; Nakagawa, K.; Narayanan, K.; Makino, S. Interplay between coronavirus, a cytoplasmic RNA virus, and nonsense-mediated mRNA decay pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 10157–10166.
- Goetz, A.E.; Wilkinson, M. Stress and the nonsense-mediated RNA decay pathway. Cell. Mol. Life Sci. 2017, 74, 3509–3531.
- Krawczak, M.; Ball, E.V.; Fenton, I.; Stenson, P.D.; Abeysinghe, S.; Thomas, N.; Cooper, D.N. Human Gene Mutation Database-A biomedical information and research resource. Hum. Mutat. 2000, 15, 45–51.
- Khajavi, M.; Inoue, K.; Lupski, J.R. Nonsense-mediated mRNA decay modulates clinical outcome of genetic disease. Eur. J. Hum. Genet. 2006, 14, 1074–1081.
- Miller, J.N.; Pearce, D.A. Nonsense-mediated decay in genetic disease: Friend or foe? Mutat. Res. Rev. Mutat. Res. 2014, 762, 52–64.
- Hall, G.W.; Thein, S. Nonsense codon mutations in the terminal exon of the β-globin gene are not associated with a reduction in β-mRNA accumulation: A mechanism for the phenotype of dominant β-thalassemia. Blood 1994, 83, 2031–2037.
- Romão, L.; Inácio, A.; Santos, S.; Avila, M.; Faustino, P.; Pacheco, P.; Lavinha, J. Nonsense mutations in the human beta-globin gene lead to unexpected levels of cytoplasmic mRNA accumulation. Blood 2000, 96, 2895–2901.
- Alrahbeni, T.; Sartor, F.; Anderson, J.; Miedzybrodzka, Z.; McCaig, C.; Müller, B. Full UPF3B function is critical for neuronal differentiation of neural stem cells. Mol. Brain 2015, 8, 33.
- Jolly, L.A.; Homan, C.C.; Jacob, R.; Barry, S.; Gecz, J. The UPF3B gene, implicated in intellectual disability, autism, ADHD and childhood onset schizophrenia regulates neural progenitor cell behaviour and neuronal outgrowth. Hum. Mol. Genet. 2013, 22, 4673–4687.
- Feng, Q.; Snider, L.; Jagannathan, S.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J.; Bradley, R.K. A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. ELife 2015, 4, e04996.
- Baradaran-Heravi, A.; Balgi, A.D.; Zimmerman, C.; Choi, K.; Shidmoossavee, F.S.; Tan, J.S.; Bergeaud, C.; Krause, A.; Flibotte, S.; Shimizu, Y. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016, 44, 6583–6598.
- Roy, B.; Leszyk, J.D.; Mangus, D.A.; Jacobson, A. Nonsense suppression by near-cognate tRNAs employs alternative base pairing at codon positions 1 and 3. Proc. Natl. Acad. Sci. USA 2015, 112, 3038–3043.
- Wilschanski, M.; Yahav, Y.; Yaacov, Y.; Blau, H.; Bentur, L.; Rivlin, J.; Kerem, E. Gentamicin-Induced Correction of CFTR Function in Patients with Cystic Fibrosis and CFTR Stop Mutations. N. Engl. J. Med. 2003, 349, 1433–1441.
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Mendell, J.R. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780.
- Peltz, S.W.; Morsy, M.; Welch, E.M.; Jacobson, A. Ataluren as an Agent for Therapeutic Nonsense Suppression. Annu. Rev. Med. 2013, 64, 407–425.
- Holbrook, J.A.; Neu-Yilik, G.; Hentze, M.W.; Kulozik, A.E. Nonsense-mediated decay approaches the clinic. Nat. Genet. 2004, 36, 801–808.
- Perrin-Vidoz, L.; Sinilnikova, O.M.; Stoppa-Lyonnet, D.; Lenoir, G.M.; Mazoyer, S. The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum. Mol. Genet. 2002, 11, 2805–2814.
- Fan, S.; Yuan, R.-Q.; Ma, Y.X.; Meng, Q.; Goldberg, I.D.; Rosen, E.M. Mutant BRCA1 genes antagonize phenotype of wild-type BRCA1. Oncogene 2001, 20, 8215–8235.
- Sasaki, C.Y.; Lin, H.; Morin, P.J.; Longo, D.L. Truncation of the Extracellular Region Abrogrates Cell Contact but Retains the Growth-suppressive Activity of E-cadherin. Cancer Res. 2000, 60, 7057–7065.
- Karam, R.; Carvalho, J.; Bruno, I.; Graziadio, C.; Senz, J.; Huntsman, D.; Oliveira, C. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene 2008, 27, 4255–4260.
- Kaurah, P.; MacMillan, A.; Boyd, N.; Senz, J.; De Luca, A.; Chun, N. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA 2007, 297, 2360–2372.
- Liu, C.; Karam, R.; Zhou, Y.; Su, F.; Ji, Y.; Li, G.; Lu, Y. The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat. Med. 2014, 20, 596–598.
- Cao, L.; Qi, L.; Zhang, L.; Song, W.; Yu, Y.; Xu, C.; Li, L.; Guo, Y.; Yang, L.; Liu, C.; et al. Human nonsense-mediated RNA decay regulates EMT by targeting the TGF-ß signaling pathway in lung adenocarcinoma. Cancer Lett. 2017, 403, 246–259.
- Gubanova, E.; Brown, B.; Ivanov, S.V.; Helleday, T.; Mills, G.B.; Yarbrough, W.G.; Issaeva, N. Downregulation of SMG-1 in HPV-positive head and neck squamous cell carcinoma due to promoter hypermethylation correlates with improved survival. Clin. Cancer Res. 2012, 18, 1257–1267.
- Mort, M.; Ivanov, D.; Cooper, D.N.; Chuzhanova, N.A. A meta-analysis of nonsense mutations causing human genetic disease. Hum. Mutat. 2008, 29, 1037–1047.