Acute myeloid leukemia (AML) is a heterogenous hematopoietic neoplasm with various genetic abnormalities in myeloid stem cells leading to differentiation arrest and accumulation of leukemic cells in bone marrow (BM). The multiple genetic alterations identified in leukemic cells at diagnosis are the mainstay of World Health Organization classification for AML and have important prognostic implications. Recently, understanding of heterogeneous and complicated molecular abnormalities of the disease could lead to the development of novel targeted therapeutic agents. In the past years, gemtuzumab ozogamicin, BCL-2 inhibitors (venetovlax), IDH 1/2 inhibitors (ivosidenib and enasidenib) FLT3 inhibitors (midostaurin, gilteritinib, and enasidenib), and hedgehog signaling pathway inhibitors (gladegib) have received US Food and Drug Administration (FDA) approval for the treatment of AML. Especially, AML patients with elderly age and/or significant comorbidities are not currently suitable for intensive chemotherapy. Thus, novel therapeutic planning including the abovementioned target therapies could lead to improve clinical outcomes in the patients.
- acute myeloid leukemia
- gemtuzumab ozogamicin
- BCL-2
1. Introduction
2. Anti-CD33 Directed Antibody
Mechanism of Action
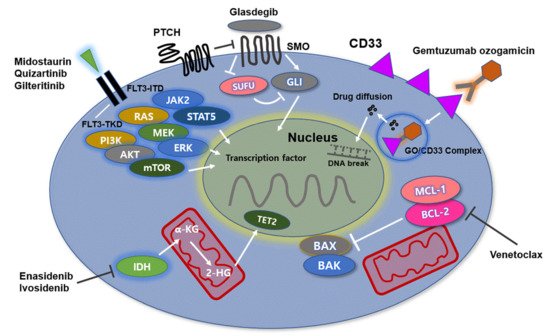
3. Gemtuzumab Ozogamicin, Anti-CD33 Antibody
3.1. Clinical Data
| Author (Refer.) | Therapeutic Schedule | Phase/Population | Clinical Outcome |
|---|---|---|---|
| Anti-CD33 monoclonal antibody | |||
| Petersdorf et al. | GO—6 mg/m2 on day 4. additional 3 doses, 5 mg/m2 in CR patients after consolidation GO + modified DA vs. standard DA |
Phase III, ND AML, n= 595 | ORR, 76% in DA plus GO group vs. 74% in DA alone (p = 0.36) CR, 69% vs. 70% (p = 0.69) 5-yr RFS, 47% vs. 42% (p = 0.87) 5-yr OS, 46% vs. 50% (p = 0.09) |
| Castaigne et al. ALFA-0701 |
DA +/− GO—3 mg/m2 for day 1, 4, and 7 of induction, 3 mg/m2 for day 1 of two consolidations |
Phase III, ND AML, n = 278 | CR/CRi, 81 in GO + group vs. 75% in GO—group (p = 0.25) 2-yr EFS, 40.8 vs. 17.1% (p = 0.0003) 2-yr OS, 53.2 vs. 41.9% (p = 0.0368) 2-yr RFS, 50.3 vs. 22.7% (p = 0.0003) Survival benefit—favorable and intermediate-risk group |
| Burnett et al. MRC-AML15 |
GO—3 mg/m2 for day 1 + DA, 2 cycles, FLAG-ida or ADE |
Phase III, ND AML, n = 1113 | Addition of GO—no different in OS, RFS, and TRM. But, OS ↑ in favorable cytogenetic risk (79 vs. 51%, p = 0.0003) |
| Burnett et al. NCRI-AML16 and LRF AML 14 |
GO—3 mg/m2 for day 1 + DA or DC (daunorubicin + claforabine, D 1-5) |
Phase III, ND AML, n = 1115 | IC—↑ RFS (28 vs. 23%, p = 0.03) and ↑ CR (35 vs. 29 and, p = 0.04) Non-IC—↑ ORR (17 vs. 30%, p = 0.006) and ↑ CR (11 vs. 21%, p = 0.002) But, no improvement of OS |
| Burnett et al. NCRI-AML17 |
GO—3 mg or 6 mg/m2 for day 1 + DA or ADE (DA + etoposide) |
Phase III, ND AML, n = 788 | Significant higher CR rate in 3 mg GO group vs. 6 mg group (p = 0.03) 6 mg group—higher 30 and 60-day TRM (p = 0.02; p = 0.01) |
| Delaunay et al. GEOLAMS-AML 2006 IR |
GO—6 mg/m2 for day 1 + DA | Phase III, ND AML, n = 238 | CR—not different between GO + vs. GO- group (91.6 vs. 86.5%, p = NS) EFS, OS—not different between GO + vs. GO- group.VOD, hepatotoxicity, higher in GO + group (23 vs. 13%; p = 0.031) |
| Burnett et al. EORTC-GIMEMA AML 19 |
GO—6 mg/m2 for day 1, 3 mg/m2 for day 8 vs. Best supportive care |
Phase III, ND AML unfit for IC, n = 237 |
OS, 4.9 months in GO group vs. 3.6 months BSC group (p = 0.005) 1-yr OS, 24.3% vs. 9.7% OS benefit of GO, higher in women and favorable, intermediate-risk group. CR + CRi in GO group, 27% |
| BCL-2 inhibitor | |||
| Combination study with hypomethylating agents | |||
| DiNardo et al. Blood 2019 |
Venetoclax, 400, 800, 1200 mg + HMAs (AZA, or DEC) | ND AML ≥ 60 years or unfit for IC, n = 145 |
CR/CRi, 67% in all patients; CR/CRi, 73% in venetoclax 400 mg/day group Median CR/CRi duration, 11.3 months Median OS, 17.5 months |
| DiNardo et al. NEJM 2020 |
Venetoclax, 400 mg/day + AZA | Phase III, ≥75 years or unfit for IC, n = 431 |
OS, 14.7 months in venetoclax-AZA group vs. 9.6 months in control (p < 0.001) CR/CRi, 36.7%/66.4% in venetoclax-AZA group vs. 17.6%/28.3% in control (p < 0.001) |
| Combination study with Low dose cytarabine | |||
| Wei et al. (JCO) | Venetoclax, 600 mg/day + LDAC | Median age, 74 yrs (range, 63–90 yrs) In enrolled patients CR/CRi, 54%; OS, 10.1 months; DOR, 8.1 months In patients without prior HMA exposure, CR/CRi, 62%; DOR, 14.8 months; OS, 13.5 months |
|
| Wei et al. (blood) | Venetoclax, from 100 mg/day to 600 mg/day + LDAC |
ND AML unfit for IC, n = 211 | Median age, 76 yrs (range, 36–93 yrs) OS, 8.4 mos in venetoclax + LDAC vs. 4.1 mos in LDAC alone (p = 0.04). CR/CRi, 48% in venetoclax + LDAC vs. 13% in LDAC alone (p < 0.001) |
| FLT3 inhibitor | |||
| Midostaurin | |||
| Stone et al. | Midostaurin, 50 mg/day twice/day + DA | Phase Ib, ND AML, n = 29 | CR, 92% in FLT3-ITD + vs. 74% in FLT3-WT (p = NS) 1 and 2-yr OS, 0.85, 0.62 in FLT3-ITD+ vs. 0.78, 0.52 in FLT3-WT (p = NS) 1-yr DFS, 50 in FLT3-ITD+ vs. 60% in FLT3-WT (p = NS) |
| Stone et al. | DA +/− Midostaurin, 50 mg/day twice/day | Phase III, ND AML, n = 717 | OS, 74.7 in midostaurin, higher than 25.6 months in placebo (p = 0.009) EFS, in midostaurin group, higher than placebo (p = 0.002) CR, 58.9 in midostaurin vs. 53.5% in placebo (p = NS). Midostaurin, beneficial in both ITD and TKD mutation Severe toxicity, similar between two groups (p = NS) |
| Quizartinib | |||
| Cortes et al. (JCO) |
quizartinib, escalating doses of 12 to 450 mg/day | Phase I, R/R AML +/− FLT3 status, n =76 |
In enrolled patients—ORR/CR—30%/13% ORR—53% in FLT3-ITD group vs. 14% FLT3-WT group |
| Cortes et al. (lancet) |
quizartinib monotherapy | Phase II cohort, R/R AML, n = 333 Cohort 1 ≥ 60 yrs, R/R within 1 yr Cohort 2 ≥ 18 yrs, R/R after salvage or SCT |
Cohort 1 Composite CR/CR—56%/3% in FLT3-ITD group Compositive CR/CR—36%/5% in FLT3-WT group Cohort 2 Composite CR/CR—46%/4% in FLT3-ITD group Compositive CR/CR—30%/3% in FLT3-WT group |
| Cortes et al. | quizartinib vs. investigator’s choice | Phase III, R/R AML with FLT-ITD +, n = 367 |
OS, 6.2 in quizartinib vs. 4.7 months in chemotherapy (p = 0.02) Therapy-related death, 17% vs. 17% (p = NS) |
| Gilteritinib | |||
| gilteritinib, 120 mg/day vs. salvage chemotherapy | Phase III, R/R AML with FLT-ITD +, n = 371 |
OS, 9.3 in gilteritinib vs. 5.6 months in chemotherapy (p < 0.001) EFS, 2.8 months vs. 0.7 months (p = NS). CR with hematologic recovery, 34.0 vs. 15.3% (18.6, 95% CI; 9.8-27.4) |
|
| IDH1/2 inhibitor | |||
| Enasidenib | |||
| Stein et al. | Dose-escalation phase, 50–650 mg/day/day Expansion phase, 100 mg/day.day |
Phase I/2, R/R AML, n= 214 |
Median age, 68 years. ORR/CR—38.8%/19.6% BMT proceeding rate—10.3% Medians OS, 8.8 months RBC/PLT transfusion independence—40.2%/43.1% |
| Klink et al. | Enasienib, 50–650 mg/day/day Control group—other treatment group |
Retrospective, R/R AML, n = 200 |
Enasidenib, less refractory to induction than control group (p = 0.02) CR/PR/LFS, enasidenib group, higher than control (p < 0.01) Median PFS, 8.4 vs. 4.8 months (p = <0.01) Median OS, 11.0 vs. 6.4 months (p < 0.01) |
| Riva et al. | Enasidenib, 100 mg/day/day Control group—other treatment group |
Retrospective, R/R AML n = 37 |
Median OS in enasidenib, higher than control (p = 0.0419) PFS (p = NS) |
| Ivosidenib | |||
| DiNardo et al. | ivosidenib 500 mg/d | Phase I, R/R AML, n = 125 | Median follow-up duration, 14.8 monthsORR/CRh/CR—41, 30, 22% Duration of ORR/CRh/CR—6.5/8.2/9.3 months In F/U 14.8 months, median OS 8,8 months |
| Paschka et al. | Ivosidenib, 500 mg/day Control group—other treatment group |
Data analysis, R/R AML, n = 434 |
OS, 8.1 in ivosidenib vs. 2.9 months control group (p < 0.0001) 6/12-month survival rate—55.7%/35.0 vs. 29.1%/10.8% (p < 0.001) CR—18.3% vs. 7.0% (p < 0.001) |
| Hedgehog signaling inhibitor | |||
| Glasdegib | |||
| Cortes et al. | Glasdegib, 100 mg + LDAC vs. LDAC alone | Phase II, ND AML unfit for IC, n = 132 |
Median OS was 8.8 months with glasdegib group vs. 4.9 months with LDAC group (p = 0.0004) CR, 17% in gladegib group vs. 2.3% in LDAC group (p < 0.05) Grade ≥ 3 AE, pneumonia (16.7%), fatigue (14.3%) |
3.2. BCL-2 Inhibitor
Mechanisms of Action
3.3. Venetoclax, BCL-2 Inhibitor
Clinical Data
References
- Medinger, M.; Passweg, J.R. Acute myeloid leukaemia genomics. Br. J. Haematol. 2017, 179, 530–542.
- Pulte, D.; Gondos, A.; Brenner, H. Expected long-term survival of patients diagnosed with acute myeloblastic leukemia during 2006–2010. Ann. Oncol. 2009, 21, 335–341.
- Appelbaum, F.R.; Gundacker, H.; Head, D.R.; Slovak, M.L.; Willman, C.L.; Godwin, J.E.; Anderson, J.E.; Petersdorf, S.H. Age and acute myeloid leukemia. Blood 2006, 107, 3481–3485.
- Chen, K.T.J.; Gilabert-Oriol, R.; Bally, M.B.; Leung, A.W.Y. Recent Treatment Advances and the Role of Nanotechnology, Combination Products, and Immunotherapy in Changing the Therapeutic Landscape of Acute Myeloid Leukemia. Pharm. Res. 2019, 36, 1–20.
- Perl, A.E. The role of targeted therapy in the management of patients with AML. Blood Adv. 2017, 1, 2281–2294.
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447.
- Herold, T.; Rothenberg-Thurley, M.; Grunwald, V.V.; Janke, H.; Goerlich, D.; Sauerland, M.C.; Konstandin, N.P.; Dufour, A.; Schneider, S.; Neusser, M.; et al. Validation and refinement of the revised 2017 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia 2020, 34, 3161–3172.
- Kantarjian, H.M.; Thomas, X.G.; Dmoszynska, A.; Wierzbowska, A.; Mazur, G.; Mayer, J.; Gau, J.-P.; Chou, W.-C.; Buckstein, R.; Cermak, J.; et al. Multicenter, Randomized, Open-Label, Phase III Trial of Decitabine Versus Patient Choice, With Physician Advice, of Either Supportive Care or Low-Dose Cytarabine for the Treatment of Older Patients with Newly Diagnosed Acute Myeloid Leukemia. J. Clin. Oncol. 2012, 30, 2670–2677.
- Dombret, H.; Seymour, J.F.; Butrym, A.; Wierzbowska, A.; Selleslag, D.; Jang, J.H.; Kumar, R.; Cavenagh, J.D.; Schuh, A.C.; Candoni, A.; et al. International phase 3 study of azacitidine vs conventional care regimens in older patients with newly diagnosed AML with >30% blasts. Blood 2015, 126, 291–299.
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjugate Chem. 2002, 13, 47–58.
- Paul, S.P.; Taylor, L.S.; Stansbury, E.K.; McVicar, D.W. Myeloid specific human CD33 is an inhibitory receptor with differential ITIM function in recruiting the phosphatases SHP-1 and SHP-2. Blood 2000, 96, 483–490.
- Appelbaum, F.R.; Bernstein, I.D. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood 2017, 130, 2373–2376.
- Sievers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: A phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood 1999, 93, 3678–3684.
- Zein, N.; Sinha, A.M.; McGahren, W.J.; A Ellestad, G. Calicheamicin gamma 1I: An antitumor antibiotic that cleaves double-stranded DNA site specifically. Science 1988, 240, 1198–1201.
- Rosen, D.B.; Harrington, K.H.; Cordeiro, J.A.; Leung, L.Y.; Putta, S.; Lacayo, N.; Laszlo, G.S.; Gudgeon, C.J.; Hogge, D.E.; Hawtin, R.E.; et al. AKT Signaling as a Novel Factor Associated with In Vitro Resistance of Human AML to Gemtuzumab Ozogamicin. PLoS ONE 2013, 8, e53518.
- Linenberger, M.L. CD33-directed therapy with gemtuzumab ozogamicin in acute myeloid leukemia: Progress in understanding cytotoxicity and potential mechanisms of drug resistance. Leukemia 2004, 19, 176–182.
- Amico, D.; Barbui, A.M.; Erba, E.; Rambaldi, A.; Introna, M.; Golay, J.T. Differential response of human acute myeloid leukemia cells to gemtuzumab ozogamicin in vitro: Role of Chk1 and Chk2 phosphorylation and caspase 3. Blood 2003, 101, 4589–4597.
- Sievers, E.L.; Larson, R.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and Safety of Gemtuzumab Ozogamicin in Patients with CD33-Positive Acute Myeloid Leukemia in First Relapse. J. Clin. Oncol. 2001, 19, 3244–3254.
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860.
- Burnett, A.K.; Russell, N.H.; Hills, R.; Hunter, A.E.; Kjeldsen, L.; Yin, J.; Gibson, B.E.; Wheatley, K.; Milligan, D. Optimization of Chemotherapy for Younger Patients with Acute Myeloid Leukemia: Results of the Medical Research Council AML15 Trial. J. Clin. Oncol. 2013, 31, 3360–3368.
- Burnett, A.K.; Hills, R.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of Patients with Acute Myeloblastic Leukemia Who Benefit from the Addition of Gemtuzumab Ozogamicin: Results of the MRC AML15 Trial. J. Clin. Oncol. 2011, 29, 369–377.
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.-N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516.
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996.
- Burnett, A.; Cavenagh, J.; Russell, N.; Hills, R.; Kell, J.; Jones, G.; Nielsen, O.J.; Khwaja, A.; Thomas, I.; Clark, R.; et al. Defining the dose of gemtuzumab ozogamicin in combination with induction chemotherapy in acute myeloid leukemia: A comparison of 3 mg/m2 with 6 mg/m2 in the NCRI AML17 Trial. Haematology 2016, 101, 724–731.
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients with Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III EORTC-GIMEMA AML-19 Trial. J. Clin. Oncol. 2016, 34, 972–979.
- Cheng, E.H.; Wei, M.C.; Weiler, S.; Flavell, R.A.; Mak, T.W.; Lindsten, T.; Korsmeyer, S.J. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell. 2001, 8, 705–711.
- Wei, A.H.; Strickland, S.A., Jr.; Hou, J.Z.; Fiedler, W.; Lin, T.L.; Walter, R.B.; Enjeti, A.; Tiong, I.S.; Savona, M.; Lee, S.; et al. Venetoclax Combined with Low-Dose Cytarabine for Previously Untreated Patients with Acute Myeloid Leukemia: Results from a Phase Ib/II Study. J. Clin. Oncol. 2019, 37, 1277–1284.
- Dinardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17.
- Bogenberger, J.M.; Delman, D.; Hansen, N.; Valdez, R.; Fauble, V.; Mesa, R.A.; Tibes, R. Ex vivo activity of BCL-2 family inhibitors ABT-199 and ABT-737 combined with 5-azacytidine in myeloid malignancies. Leuk. Lymphoma 2015, 56, 226–229.
- Tsao, T.; Shi, Y.; Kornblau, S.; Lu, H.; Konoplev, S.; Antony, A.; Ruvolo, V.; Qiu, Y.H.; Zhang, N.; Coombes, K.R.; et al. Concomitant inhibition of DNA methyltransferase and BCL-2 protein function synergistically induce mitochondrial apoptosis in acute myelogenous leukemia cells. Ann. Hematol. 2012, 91, 1861–1870.
- Dinardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629.
- Wei, A.H.; Montesinos, P.; Ivanov, V.; Dinardo, C.D.; Novak, J.; Laribi, K.; Kim, I.; Stevens, D.A.; Fiedler, W.; Pagoni, M.; et al. Venetoclax plus LDAC for newly diagnosed AML ineligible for intensive chemotherapy: A phase 3 randomized placebo-controlled trial. Blood 2020, 135, 2137–2145.