Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS) is a neurodegenerative disorder seen in older premutation (55–200 CGG repeats) carriers of FMR1. Three molecular mechanisms have been proposed: (1) the production of toxic FMRpolyG by repeat-associated non-AUG (RAN) translation, (2) RNAs and protein sequestration into intranuclear inclusions and (3) DNA damage caused by R-loop formation. Additionally, mitochondrial dysfunction, oxidative stress, and iron metabolism dysregulation also play a role in the development of the neuropathology of FXTAS. One of the neuropathological hallmarks of FXTAS is the presence of eosinophilic intranuclear inclusions in the CNS and peripheral nervous system. Other findings include cerebral atrophy, ventriculomegaly, and spongiosis of the cerebellar white matter. The mechanisms underlying the pathophysiology of FXTAS are currently being investigated for the development of targeted treatment.
- FXTAS
- RANT
- Pathophysiology
- FMR1
- Protein sequestration
- Intranuclear inclusions
- Iron accumulation
- Oxidative stress
- Microglia
- mitochondrial dysfunction
1. Molecular Mechanisms
The neurotoxic effect of increased levels of FMR1 mRNA has been proposed as the main alteration leading to the development of Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS)[1] [12]; increased levels of mRNA also cause Ca+2 dysregulation followed by mitochondrial dysfunction[2][3] [119,120]. Three different molecular mechanisms have been studied with the aim of understanding the molecular abnormalities resulting in the neuropathology of FXTAS: (1) the production of toxic FMRpolyG by repeat associated non-AUG (RAN) translation, (2) RNAs and protein sequestration into intranuclear inclusions and (3) DNA damage caused by R-loop formation.
The first proposed mechanism is RAN translation[4] [121]. RAN translation is common in triplet expansion disorders. This error in translation leads to anomalous peptide synthesis[4][5][6][7][8] [121–125]. Particularly in FXTAS, the noncoding region of FMR1 premutation mRNA is translated into multiple RAN translation peptides, as seen in Figure 1, including the FMRpolyG peptide[4][9] [121,126]. This peptide has been found to be toxic by disrupting the architecture of the nuclear lamina due to its interaction with the transmembrane protein lamina-associated polypeptide 2 beta (LAP2β) in differentiated neurons derived from FXTAS induced pluripotent stem (iPS) cells. The length of FMRpolyG correlates with the number of CGG repeats; it is detected in CGG expansions ranging from 60 to 200 repeats and it is also present in the intranuclear inclusions pathognomonic of FXTAS in the FMR1 premutation transgenic animal models[9] [126].
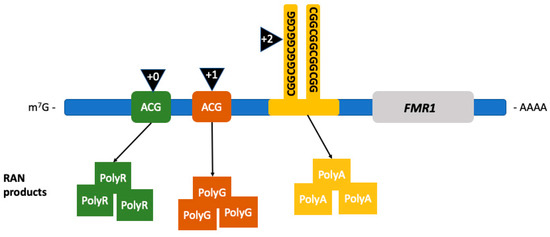
Figure 1. Repeat associated non-AUG (RAN) translation in FXTAS. RAN translation leads to the production of RAN products depending on the location where the initiation of translation occurs. Initiation (black triangles) occurring in the +0 reading frame leads to the production of FMRpolyR (green squares), +1 to FMRpolyG (brown squares), and +2, within the CGG-repeat region, to the production of FMRpolyA (yellow squares). (Adapted from Glineburg et al. 2018)[10] [127].
The second mechanism is the sequestration of RNA and other proteins by the FMR1 premutation mRNA forming intranuclear inclusions, disrupting essential cellular processes. Protein sequestration renders the cell functionally deficient from proteins that play an important role in multiple cellular mechanisms such as splicing (heterogeneous nuclear ribonucleoproteins A2/B1 protein (hnRNP A2/B1)), mRNA transportation in the cytoplasm (hnRNP A2/B1, Pur-alpha), microRNA processing (DiGeorge Syndrome Critical Region 8 (DGCR8) protein and Drosha complex), and formation of heterochromatin (heterochromatin protein 1 (HP1))[10][11] [127,128]. Figure 2 shows the proteins involved in FMR1 RNA-induced sequestration. Ubiquitin positive inclusions are the hallmark of FXTAS[12][13][14] [75,129,130]; they contain FMR1 mRNA but lack FMRP[15][16] [131,132]. Inclusions are an aggregate of protein, composed of a heterogeneous assortment of many peptides. The DNA damage response proteins found in the inclusions form in response to oxidative stress leading to neurodegeneration[16] [132]. The mechanisms by which protein sequestration leads to toxicity and disease are currently being studied.
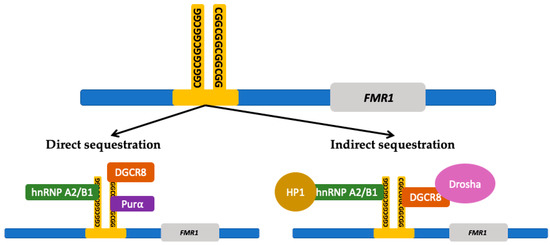
Figure 2. FMR1 RNA induced protein sequestration. The expanded CGG-repeat region in the FMR1 free mRNA can form higher-order structures that can sequester proteins which results in a deficiency within the cell from those proteins. The CGG-repeat region is directly bound by repeat binding proteins such as heterogeneous nuclear ribonucleoproteins A2/B1 (hnRNP A2/B1), DiGeorge syndrome critical region 8 (DGCR8) protein, and Pur-alpha ((Purα) protein. The CGG-repeat region can also indirectly sequester other proteins through the interaction with the directly bound proteins. Proteins that can be indirectly sequestered include Drosha and heterochromatin protein 1 (HP1). (Adapted from Hagerman and Hagerman 2016 and Glineburg et al. 2018)[1][10] [12,127].
The third proposed molecular mechanism involves the formation of RNA/DNA hybrids or R-loops during transcription. R-loops form during the CGG triplet expansion at the FMR1 locus causing vulnerability to DNA damage[17] [133]. These R-loops are more frequently formed in the nontemplate strand containing guanine-rich sequences[18][19] [134,135]. These folded structures remain unpaired and lead to genome instability[20] [136]. The sites become fragile and are prone to DNA damage including deletions and translocations. DNA damage should be corrected by the DNA damage response (DDR) molecular signaling pathway[21] [137]. However, this response appears to be impaired in FXTAS[22] [138].
The molecular mechanisms presented here do not exclude one another; on the contrary, there may exist a synergistic effect from different dysfunctional processes at the molecular level leading to FXTAS. The mitochondrial abnormalities present in FXTAS may worsen over time and lead to loss of energy and strength in the patients and perhaps neuronal cell death and white matter disease over time[23][24][25][26] [119,139–141]. Many additional mechanisms causing neuropathology and toxicity may remain unrecognized.
2. Pathology
One of the neuropathological hallmarks of FXTAS is the presence of eosinophilic intranuclear inclusions in the CNS and peripheral nervous system[27][28] [22,84]. The presence of inclusions in neurons and astrocytes constitutes a major criterion for the diagnosis of FXTAS[27][29] [22,142]. These intranuclear inclusions were first identified in neurons and astrocytes in the brain in 2002 by Greco and colleagues[13] [129]. Inclusions occur broadly throughout the CNS with regional variability in the proportion of cells bearing the inclusions[30] [143]. Neurons and astrocytes positive for the presence of inclusions are mostly prevalent in the hippocampus (up to 40% of cells), followed by the frontal and the temporal cortex[31][12][13] [67,75,129]. These inclusions have also been identified in the basal ganglia[32] [97]. Further studies identified these inclusions in the peripheral nervous system, mainly in the autonomic system[33] [48], and other non-CNS locations such as neuroendocrine tissue, heart, and kidney[28][32][34] [84,97,144].
Various studies have been conducted in order to elucidate the features of the inclusions. These eosinophilic inclusions have shown to stain positive for ubiquitin, lamin A/C and various heat-shock proteins, and their biochemical composition is very heterogeneous with no single predominant protein species[15] [131]. Mass spectrometric and immunohistochemical analysis in postmortem FXTAS brain tissue have identified more than 20 inclusion-associated proteins including RNA-binding protein, hnRNP A2, intermediate filament proteins, and other neurofilament proteins[12][13][15] [75,129,131]. FMR1 mRNA was also identified but as a minor component[35] [145]. More recently, Ma and colleagues conducted a study of the composition of the intranuclear inclusions of FXTAS using fluorescence-activated cell sorting (FACS) and liquid chromatography/tandem mass spectrometry-based proteomics. They identified more than 200 proteins which are normally involved in RNA binding, protein turnover, and DNA damage repair[16] [132]. It is noteworthy that the presence of intranuclear inclusions has also been identified in a carrier with no clinical symptoms associated with FXTAS, although this carrier was bedridden with severe ataxia and perhaps other symptoms associated with FXTAS by the time she died, so it is uncertain if the presence of inclusions always means FXTAS, but this is likely the case[31] [67].
Iron metabolic pathway dysregulation has been identified as a mechanism underlying the neuropathology of FXTAS and therefore as a potential targeted treatment. Postmortem FXTAS brains present with iron accumulation in brain capillaries and parenchyma, as well as in the choroid plexus[36][37][38] [146–148]. Severe iron accumulation is a common finding in the striatum and less commonly in the cerebrum[36] [146]. There is a decreased amount of transferrin and ceruloplasmin, which are iron-binding proteins, in neurons and astrocytes, whereas there are increased levels of these proteins in the microglial cells, which indicates an attempt to respond to excessive iron accumulation[36] [146]. Studies have demonstrated substantial iron bound to hemosiderin accumulated in the putamen[36][37][39] [146,147,149], which on neuroimaging can be detected as symmetric hypointensities in the putamen and caudate in T2-weighted MRI[40][41] [85,114].
Since FXTAS is associated with high levels of oxidative stress in the brain and an inflammatory state, and considering that microglia-mediated neuroinflammation plays a major role in the pathogenesis of neurodegenerative diseases, it was proposed that microglial activation could contribute to FXTAS pathology[42] [150]. In a study of 13 FXTAS brain specimens, Martínez-Cerdeño and colleagues found that almost half of FXTAS brains presented with dystrophic senescent microglial cells, suggesting that these could be used in association with the presence of intranuclear inclusions and iron deposits as a marker in the postmortem diagnosis of FXTAS[42] [150]. They also found that the presence of senescent microglia was correlated with the CGG repeat number and with iron accumulation. This suggests that microglial cells are involved in the neuroinflammatory state of FXTAS and that they can serve as a pathological criterion for the diagnosis.
FXTAS can coexist with other neurodegenerative diseases. Salcedo-Arellano and colleagues conducted a study to describe the frequency of Parkinson’s disease and concomitant FXTAS[43] [151]. They reviewed medical records and performed a pathology analysis on 40 deceased patients with a diagnosis of FXTAS. They found that 5% of the brains of 40 patients met pathologic criteria for both Parkinson’s disease, based on the presence of Lewy bodies in the substantia nigra and nigral neuronal loss, as well as FXTAS. In the case of Alzheimer’s disease, some patients with FXTAS and dementia symptomology presented with concurrent findings of Alzheimer’s neuropathology such as amyloid plaques and neurofibrillary tangles[31] [67]. Various case reports of patients with FXTAS and coexisting Alzheimer’s disease or Parkinson’s disease suggest a synergistic effect on the progression of FXTAS and the concomitant neurodegenerative disease[31][44][45] [67,152,153].
Finally, macroscopic pathological findings of brains from patients with FXTAS reveal severe white matter disease, cortical atrophy and mild to severe ventriculomegaly and spongiosis of cerebellar white matter[31][12][13] [67,75,129]. Grey matter atrophy is most consistently found in the frontal cortex, cerebellum, and the pons[12][13] [75,129].