Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Eric Toth and Version 2 by Rita Xu.
The envelope of HCV contains two glycoproteins, E1 and E2, that are membrane-anchored via their transmembrane domains and heterodimerize as the E1E2 complex, which in turn further multimerizes to form a trimer of heterodimers on the surface of the virion. The E1E2 complex governs viral entry and is the primary target of protective antibodies.
- hepatitis C virus (HCV)
- envelope glycoproteins
- E1
- E2
- E1E2 glycoprotein complex
1. HCV E1E2 and Vaccine Development
The global burden of hepatitis C virus (HCV) infection is about 71 million or approximately 1% of the world’s population, with an annual rate of 1.75 million new infections each year [1]. Chronic HCV infection can lead to cirrhosis and hepatocellular carcinoma, which is the most common type of primary liver cancer [2]. The development of direct-acting antivirals has improved treatment options considerably, but their high cost restricts access, particularly in developing countries where the disease burden is greatest. Moreover, other factors such as viral resistance, the occurrence of reinfections after treatment cessation, and the fact that HCV is largely asymptomatic until the infected person sustains significant liver damage can diminish the impact of antiviral treatments. Therefore, the development of an effective preventative vaccine for HCV is imperative to lessen the burden of infection and transmission and to eliminate HCV globally [3].
Cumulative evidence has established that both B- and T-cell immunity contribute to the control of acute HCV infection [4][5][6][4,5,6]. In particular, broadly neutralizing antibodies (bnAbs) infused into humanized mice or chimpanzees protect against HCV infection [7][8][9][7,8,9]. Moreover, viral clearance is associated with a robust induction of bnAbs early in the infection [10][11][12][13][10,11,12,13]. Any protective HCV vaccine will likely require an immunogen that robustly elicits bnAbs. A major challenge is a high variability in the viral envelope E1E2 glycoprotein complex, the natural target of protective antibodies [14][15][14,15]. An ideal vaccine should elicit sufficient titers of bnAbs to multiple conserved E1E2 epitopes [8][16][17][18][19][8,16,17,18,19], in conjunction with cytotoxic and tissue-resident memory T-cells to achieve immunity, ideally sterilizing immunity, and protection against a high diversity of HCV isolates.
2. Importance of E1, E2, and E1E2 Epitopes In Virus Neutralization
1.1. Importance of E1, E2, and E1E2 Epitopes In Virus Neutralization
HCV envelope glycoproteins form a trimer of heterodimers, an assembly driven by their transmembrane domains (TMDs). The envelope of HCV contains two glycoproteins, E1 and E2 that are encoded as part of the HCV polyprotein expressed in infected liver cells. This polyprotein is processed in the endoplasmic reticulum (ER) by signal peptidases and cellular glycosylation machinery to produce the mature E1E2 complex. These glycoproteins are membrane-anchored via their C-terminal helices resulting in a membrane-bound E1E2 (mbE1E2) complex. Biochemical evidence supports the observation that the E1E2 heterodimer (see Figure 2) forms a trimer on the virion. First, sedimentation analysis shows that E1E2 contained in viral particles migrates consistent with a trimeric assembly that interacts with the HCV co-receptor CD81. Second, the E1 protein migrates primarily as SDS-resistant trimers on an SDS-PAGE after mild heating. In addition, studies using both chimeric E1E2 glycoproteins with strain-swapped ectodomain-TMD combinations and studies using point mutants showed that residues in the E1 and E2 TMDs impact heterodimer assembly, incorporation into pseudoparticles, and infectivity. Moreover, mutations in the GxxxG motif in the E1 TMD eliminate the SDS-resistant trimers and the E1 TMD alone can form SDS-resistant trimers when fused to monomeric thioredoxin. Thus, the E1 TMD appears to be a key driver of E1E2 assembly.
In developing a prophylactic vaccine for HCV, the choice of antigen will be critical for its success. The HCV envelope glycoprotein complex E1E2, as the target of the protective antibody response, is currently the prime candidate. A soluble version of the E2 protein (sE2) lacking the transmembrane domain [20] has been the focus of a number of vaccine studies [21][22][23][24][25][26][27][21,22,23,24,25,26,27]. However, immunological assessment in chimpanzees of an E1E2 vaccine produced superior immune responses compared to E2 administered alone and resulted in sterilizing immunity against a homologous virus challenge [28][29][28,29] but with some cross-neutralization capacity against heterologous isolates [30]. Moreover, E1E2 has been tested in humans and is well-tolerated [31]. However, due to the limited neutralization breadth observed in the human clinical trial [32][33][32,33], ongoing research efforts are now focused on developing an optimized, modified form of E1E2 (mE1E2) that can elicit more robust, cross-neutralizing responses against multiple heterologous isolates [25][34][35][36][25,34,35,36].
To develop a vaccine for HCV, the candidate antigen needs to be produced at a large scale, reproducibly, and at a cost that is not prohibitive. Most antibodies with broad virus-neutralizing (Vn) activities to diverse HCV isolates recognize conformational epitopes in E2 and some in E1E2 [8][37][38][39][40][41][42][43][8,37,38,39,40,41,42,43]. In addition, studies with a panel of E1-specific mAbs have shown that E1-specific responses can both protect against virus challenge and broadly neutralize HCV pseudoparticles (HCVpp) of various genotypes [44][45][44,45]. Previous work has identified highly conserved antigenic regions on E2 that elicit bnAbs [43][46][47][43,46,47]. These regions are called antigenic domains B, D, and E (used here). Other groups have designated such regions as epitopes I–III [48] or antigenic regions (ARs, which include epitopes that contain residues from both E1 and E2 [8][39][8,39]). Domains B and D are conformation-sensitive, whereas domain E contains a linear epitope [46]. However, while not conformation-sensitive in the same sense as domain B and D NAbs, domain E adopts multiple conformations, as observed when bound to different anti-domain E mAbs [49][50][51][52][49,50,51,52].
3. Structural Characterization of E2 Antigens and a Model of the E1E2 Heterodimer
1.2. Structural Characterization of E2 Antigens and a Model of the E1E2 Heterodimer
Limited high-resolution structural data are available for the E1 glycoprotein. The E1 ectodomain contains 158 residues (residues 192–349 of the H77 sequence; NCBI Refseq ID NC_004102), of which 8 are cysteine residues that form 4 putative disulfide bonds and 5 are N-glycosylation sites. The reported X-ray structure of an N-terminal fragment of E1 (residues 192–270) exhibits “unexpected” domain-swapped and disulfide cross-linked oligomers [53], and it is a possibility that the observed structure and oligomerization were influenced by the C-terminal residue truncation, lack of E2 context, or both. As noted in a recent review [54], smaller, primarily helical fragments of E1 have also been structurally characterized (Figure 1).
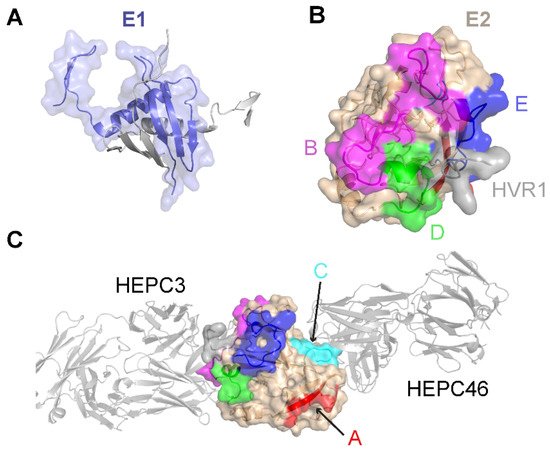
Figure 1. Structures of the E1 and E2 glycoproteins. (A) The structure of a truncated E1 glycoprotein ectodomain (PDB: 4UOI), with resolved residues encompassing positions 192–270. An E1 monomer (slate; surface and cartoon representation) is shown with a second E1 monomer (gray) representing the dimeric partner from the X-ray structure assembly. (B) The truncated E2 glycoprotein ectodomain structure (PDB: 6MEJ), encompassing E2 residues 405–645, showing the E2 neutralizing face and antigenic domains B, D, and E. Representative residues from the antigenic domains are colored as indicated by the letters, and resolved residues from hypervariable region 1 (HVR1) (residues 405–410) are colored gray. Other residues in E2 are colored tan. (C) The same E2 glycoprotein structure as in (B), reoriented to show antigenic domains A and C (colored by representative residues and labeled). Bound antibodies HEPC3 (antigenic domain B antibody) and HEPC46 (antigenic domain C antibody), both in gray cartoon, are shown for reference. The neutralizing face is colored as in panel (B).
The E2 glycoprotein has been structurally characterized more extensively than the E1 glycoprotein. The initially reported structures of the core E2 ectodomain structure [35][55][35,55] revealed a globular architecture stabilized by numerous disulfide bonds, which is expected due to the 18 cysteine residues present in the E2 ectodomain. Of note, different disulfide bonding patterns were observed in the two E2 core structures, possibly reflective of disulfide bonding heterogeneity on the native viral envelope or different truncations or source genotypes of the E2 sequences used in the two structures [56]. Subsequent studies have reported structures of E2 in complex with various monoclonal antibodies from humans [57][58][59][60][57,58,59,60] and, recently, macaques [61]. These include antibodies that engage the “neutralizing face” of E2 (Figure 1) that is used to bind the CD81 receptor, and while they exhibit different engagement strategies, many highlight the structural and mechanistic basis of shared features, such as biased VH1-69 germline gene usage and a CDRH3 loop disulfide motif [57][58][57,58]. Recent studies have provided insights into the flexibility and dynamics of the E2 neutralizing face through experimental characterization and computational modeling [36][62][63][36,62,63], suggesting possible “closed” and “open” E2 (or E1E2) conformational states associated with coreceptor binding and antibody neutralization. Analogous states and metastable behavior have been confirmed for other viral envelope glycoproteins, including HIV Env [64] and the SARS-CoV-2 spike [65].
The structure of the E1E2 glycoprotein, which would provide key insights into HCV and its immune recognition and enable structure-based vaccine design efforts, has not been experimentally determined to date. To address this need, independent studies have proposed multiple models of the E1E2 structure [66][67][66,67]; however, the lack of agreement between the E1E2 models [54], as well as inconsistencies between the initial computational models of E2 [68][69][68,69] and the E2 structure reported later, highlights the need for additional computational modeling of E1E2 in the absence of a high-resolution experimentally determined structure of this assembly. Advanced computational approaches such as the recently described artificial intelligence-based structure prediction algorithm, AlphaFold2, which was highly successful in a recent CASP13 structure prediction experiment [70], represent a promising avenue for accurate computational E1E2 modeling.
Structural data are essential for informing vaccine design, and the lack of E1E2 structural data represents a major deficit in the HCV vaccine development field. These data will aid in the optimization of neutralizing epitope presentation, engineering the antigen to make it more uniform, and enhancing stability. E1E2 is a complex antigen, so a rigorous biophysical analysis is an important part of the workflow for determining the optimal candidate.
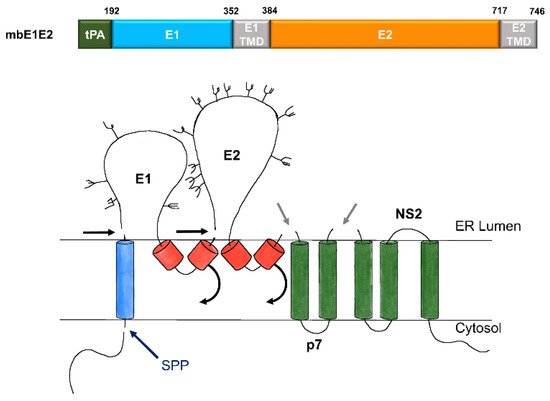
Figure 2. Expression and processing of mbE1E2. (top) Schematic of the E1E2 signal peptide (SP) plus polyprotein expression construct. (bottom) Processing pathway for the N-terminal portion of the HCV polyprotein. Signal peptidase cleavages release E1 and E2 (black and gray arrows). Signal peptide peptidase (SPP) cleavage of HCV core is indicated by a dark blue arrow. Repositioning of E1 and E2 transmembrane domains (TMDs) is indicated by curved arrows. Glycans attached to E1 and E2 are depicted as branched structures. Adapted from [71][74].