Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Aline Rangel-Pozzo and Version 2 by Bruce Ren.
Chromosome instability (CIN) is an increased rate where chromosome acquire alterations due to errors in cell division. CIN creates genetic and cytogenetic diversity and is a common feature in hematological malignancies such as acute myeloid leukemia (AML). Low to moderate levels of CIN seems to be well tolerated and can promote cancer proliferation, genetic diversity, and tumor evolution. However, high levels of CIN seems to be lethal, where enhancing CIN could improve AML treatment. However, little is known about CIN in AML.
- chromosomal instability
- acute myeloid leukemia
- cytogenetic heterogeneity
- aneuploidy
- complex karyotype
- TP53
- centrosome dysfunction
- MYC
- telomere dysfunction
- therapeutic targets
- aging
- synthetic lethality
Note: The following contents are extract from your paper. The entry will be online only after author check and submit it.
1. Introduction
Since Boveri’s theory that chromosome abnormalities promote cancer, studies have attempted to elucidate the mechanisms behind the origins of chromosomal aberrations [1]. Chromosomal instability (CIN) is the increasing rate in which cells acquire new chromosomal alterations. Depending on the type of abnormalities, it can be classified into numerical CIN (nCIN), characterized by chromosome gains and losses, and structural CIN (sCIN) represented by chromosome translocations [2]. Importantly, CIN is one of the cancer hallmarks [3]. CIN can promote selective advantage to cancer cells by increasing the probability of novel chromosomal abnormalities, which can change the expression profile of the genes regulating cell division and differentiation, resulting in high proliferation rates [3][4][3,4]. Recent studies have shown a deep relationship of CIN with the origin, progression, and relapse in many cancers [5][6][7][8][5,6,7,8].
CIN not only occurs as a tumor-promotor mechanism but also as a tumor-suppressor mechanism. This observation comes from the evidence showing that different levels of CIN lead to distinct outcomes. Moderate or low levels of CIN are associated with increased rates of genetic cancer-promoting features. On the other hand, extreme levels of CIN could lead to decreased cell fitness or apoptosis [9]. The levels of CIN and the sites in which it occurs can also indicate different outcomes [10]. Therefore, CIN features not only could refine risk stratifications but also opens opportunities for new therapeutic approaches in cancer [9][11][12][9,11,12].
The current models for CIN involve telomere dysfunction, defective spindle assembly, sister chromatid cohesion, DNA double-strand breaks (DSB) repair, genes involved in the cell cycle, and epigenetic regulators. These CIN mechanisms and their signatures can be largely found in acute myeloid leukemia (AML), a heterogeneous disease characterized by abnormal proliferation and accumulation of myeloid precursor cells in the bone marrow [13]. AML can be classified as de novo AML, secondary AML (s-AML), whose origin is from a prior hematologic disease, and therapy-related AML (t-AML), which arises as a result of exposure to alkalizing agents, irradiation, and other factors associated to prior therapy [14][15][14,15]. Regardless of the classification, approximately 55% of AML patients show chromosomal abnormalities [16]. Cytogenetic abnormalities in AML are an important prognostic factor and are used for risk-stratification and guide treatment definition [17][18][17,18]. For example, a complex karyotype (CK) is associated with poor prognosis [19]. In older patients (≥60 years), only 10–44% of those with ≥3 cytogenetic abnormalities achieve complete remission (CR) after therapy, and for those with ≥5 chromosome abnormalities, the CR rates are significantly lower (7–26%) [20][21][22][23][24][25][20,21,22,23,24,25]. In this review, we will focus on the mechanisms associated with CIN resulting in cytogenetic abnormalities (Summarized in Figure 1), their prognostic impact, and the use of CIN as a target among the AML types.
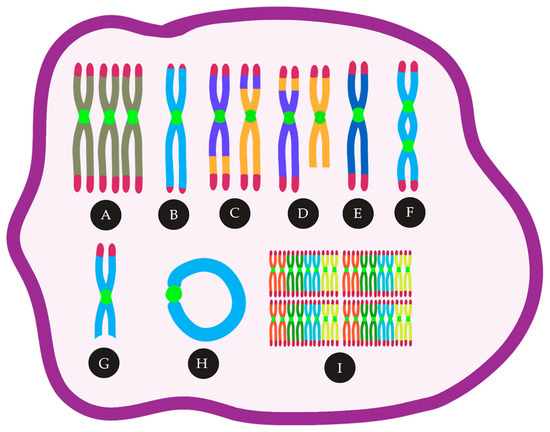
Figure 1. CIN in AML can lead to many cytogenetic abnormalities, such as (A) trisomies, (B) telomere loss, (C) reciprocal translocations, (D) unbalanced translocations, (E) monosomies, (F) Dicentric chromosomes, (G) deletions, (H) ring chromosomes, (I) polyploidy.
2. Mechanisms and Consequences of CIN
2.1. Aneuploidy and CIN in AML, Mechanism and Consequence — An Inter-Relationship
One type of nCIN is aneuploidy, characterized by an abnormal number of chromosomes in the cell, which is a result of chromosome mis-segregation errors [26]. These errors can occur when the chromosomes fail to attach correctly to the mitotic spindle [27]. CIN and aneuploidy are not synonymous [28]. Although CIN is defined as the increasing rate in which cells acquire new chromosomal alterations, aneuploidy is associated to the abnormal number of chromosomes in the karyotype at a specific point in time [29]. Aneuploidy can also be found after clonal expansion, and this is not implying that the same cell will acquire new chromosomal alterations in every cell division. That is the case of constitutional trisomies, such as trisomy 21 or Down’s syndrome, which presents aneuploidy but not CIN [30]. However, patients with trisomy of the 21 have a predisposition to cancer, such as hematologic malignancies [31]. In AML, aneuploidy is present in more than 20% of cases [17][32][33][34][17,32,33,34] and is related to poor prognosis [17][33][35][36][37][17,33,35,36,37].
A key gene to maintain diploid karyotype in normal cells is TP53 [38]. This protein regulates cellular differentiation, cell cycle arrest, and DNA repair preventing genomic instability [39][40][39,40]. Mutations in TP53 arise before or after the first aneuploidy event [41][42][41,42] These mutations are also considered an early leukemogenic event in preleukemic stem cells [43][44][43,44]. Alteration in TP53 may result in the continued proliferation of aneuploidy cells or can trigger off apoptosis [45]. TP53 has a role in suppressing nCIN and sCIN by inducing apoptosis in cells that have a long pause in the mitotic checkpoint, which indicates DNA damage [46]. Mutations in tumor suppressor genes comprise 16% of AML patients, and TP53 is one of them [34]. The incidence of TP53 mutations in AML varies between 10% and 15% [34][47][48][34,47,48]. In general, TP53 mutations are highly present in AML patients with CK (60%) [47][49][50][51][47,49,50,51].
Interesting, the incidence of TP53 mutations in AML is low in comparison with other cancer types. This observation highlights that additional mechanisms are affecting p53 function for AML patients [52]. Many pathways have been proposed for non-mutational wtp53 inactivation in AML. Mdm4, a p53 negative regulator, is overexpressed in 10% of cases in wtp53 AML with CK [53][54][53,54]. Li et al. (2014) described that Mdm4 overexpression was associated with aneuploidy or polyploidy, showing an important link between Mdm4 overexpression, wtp53 inhibition and CIN in AML [53][54][53,54]. Additionally, Mdm2 overexpression, another p53 negative regulator, is seen in more than 50% of wtp53 AML patients [55][56][57][55,56,57]. Together with Mdm2 and Mdm4 overexpression, ARF down-regulation, deregulated post-translational modifications, and nuclear-cytoplasmic microRNAs were also described as non-mutational wtp53 inactivation in AML [52]. Cazzola et al. (2019) demonstrated that AML cells TP53-knockout have CIN phenotype and karyotype heterogeneity [58]. The deletion of 5q, a chromosomal region containing many protein-encoding genes associated with hematopoietic differentiation, together with TP53 mutations, can promote genome and chromosomal instability to block normal hematopoietic differentiation and is significantly associated with CK and poor prognostic [59][60][61][59,60,61]. In addition, 5q deletion, which leads to the haploinsufficiency of the genes involved in cell cycle control, as a sole chromosomal abnormality, is rare in AML patients. However, together with TP53, this alteration is frequently found in AML patients with CK (60–69%) [62][63][64][62,63,64]. Together, these observations show a strong association between CK and the deletion of checkpoint genes (TP53 and the ones located at 5q), suggesting an important role of dysfunctional cell cycle checkpoint in AML.
2.2. Chromosome Segregation Errors
2.2.1. Defects in the Spindle Assembly Checkpoint (SAC)
Jin and Burkard (2018) have associated CIN in AML patients with defects in the spindle assembly checkpoint (SAC). SAC is a mitotic checkpoint mechanism used to prevent transition to anaphase when there is an error on the kinetochore-microtubules attachments [65][66][65,66]. When SAC is malfunctioning, cells without proper spindle attachments can bypass anaphase checkpoints and divide [1]. The inactivation of the entire mitotic checkpoint can generate chromosome mis-segregation leading to CIN or cell death [67][68][67,68] in various cancers [69][70][71][69,70,71].
Mosaic variegated aneuploidy (MVA) is a rare human chromosomal instability disorder. Patients with MVA present germline mutations in the mitotic checkpoint components and 25% of the cells show aneuploidy [72]. The most relevant mutation is in the spindle checkpoint gene BUB1B, where the SAC protein BubR1 is expressed. BubR1 stabilizes the kinetochore-microtubule and corrects the proper chromosome positioning. It prevents cell division until forming an appropriate bi-oriented mitotic spindle [73]. In MVA, mutations in BUB1B leads to CIN with consecutive constitutional mosaicism for chromosomal gains and losses and subsequent predisposition to several types of cancer [74]. In AML, Schnerch et al. (2012) suggested a role of SAC insufficiency in the pathogenesis and progression of patients with a CK [75]. Since BUBR1 is an anaphase-promoting complex (APC/C) inhibitor gene, AML cells with defects in this gene usually allow chromosomal alterations to bypass mitosis [76].
The most common chromosomal abnormality found in AML is t(8;21)(q22;q22) [77]. Boyapati et al. (2007) demonstrated in cell lines that the resulting fusion protein t(8;21) from AML impairs the spindle checkpoint and promotes aneuploidy [78]. Nucleoporin 98 gene (NUP98) is another gene associated with SAC defects in AML. NUP98 regulates the timely destruction of securin by APC/C [79]. Cells with NUP98 translocation contain aberrant securin (a regulatory protein of the metaphase-anaphase transition), leading to aneuploidy [80]. Another APC/C protein with decreased expression in AML is Cdh1, an antagonist regulator of SAC (which activates and mediates securin degradation). Therefore, the high number of SAC alterations in AML cells can be associated with other dysfunctional chromosome segregation features.
2.2.2. Cohesion Defects
Sister chromatids are kept together until the proper formation of the bipolar spindle. If cohesion between sister chromatids is lost, chromosomes mis-segregate [81]. Cohesin defects, which is a protein complex mediating sister chromatid cohesion, are also associated with aneuploidy and CIN [82][83][84][85][82,83,84,85]. Ley et al. (2013), using whole-exome sequencing, showed mutations in cohesin complex genes in 13% of de novo AML patients [34]. However, cohesin gene mutations did not show a prognostic impact in AML. This is probably due to the co-existence of cohesin complex mutations with NPM1 (Nucleophosmin 1) mutations [86]. Interestingly, the co-existence of both mutations is associated with a favorable prognosis and normal karyotype in AML [87][88][87,88]. NPM1 mutation results in the inactivation of the nuclear factor-κB (NF-κB) in the cytoplasm [89]. Since activation of NF-κB provides drug resistance to chemotherapy drugs in AML, the association between cohesion and NPM1 mutations leads to favorable prognostic and chemosensitivity.
2.2.3. Centrosome Dysfunction and Assembly of Multipolar Mitotic Spindles
Another important mechanism related to CIN is centrosome dysfunction [90][91][92][93][94][90,91,92,93,94]. Dysfunctional centrosomes are characterized by the presence of aberrant centrosomes numbers, imbalances in centrosome-associated proteins expression, centrosome structural abnormalities, and alterations in the clustering of centrosomal components [95]. Cells presenting centrosome defects show the formation of multipolar mitotic spindles (cells with multiple centrosomes) [91][96][91,96]. Neben et al. (2003) have shown an association between abnormal centrosomes and the presence of cytogenetic alterations in AML. Interestingly, centrosome dysfunction allowed stratification into cytogenetic risk groups, where higher numbers of centrosome alterations were related to an increased adverse prognosis. The authors also suggested centrosome aberrations and multipolar mitotic spindles as the cause of numerical chromosome alterations in AML patients [97].
The aurora kinases, a family of serine-threonine protein kinases, have a key role in centrosome dynamics, mitotic spindle, and mitotic centrosomes [98]. Two common types of these proteins are Aurora A and Aurora B. Aurora A is active during the late S and early G2 phase and ensures a proper spindle assembly and chromosome alignment during mitosis [99]. On the other hand, Aurora B functions as a protein complex (through the G2 phase) and is in charge of bipolar attachment of the spindle to the centromeres and correct segregation of the sister chromatids [98]. Aurora kinases A and B are overexpressed in AML CD34+ blast cells compared to CD34+ from normal individuals with no evidence of hematologic diseases [100][101][102][100,101,102]. Lucena-Araujo et al.(2010) reported that high expression of Aurora Kinases A and B was related to unfavorable cytogenetic abnormalities, represented by CK and high blasts count in AML patients [103]. Yang et al. (2013) outlined that AML blasts overexpressing Aurora A were chemotherapy resistant. Aurora A negatively regulates p53 and, due to the role of p53 in the induction of apoptosis, its downregulation allows cells to escape from apoptosis induced by chemotherapy in AML [100].
2.3. DNA Double-strand Breaks
DSBs can lead to translocations and DNA deletions [104][105][106][104,105,106]. One of the major causes of DSBs is a failure in the chromosome decatenation (disentanglement of the chromosomes) [107][108][109][107,108,109]. In normal cells, the decatenation checkpoint during the G2 phase delays the entry into mitosis until every chromosome is decatenated by the enzyme topoisomerase IIα (topo II) [109][110][109,110]. Wray et al. (2009) reported that AML cells that fail to arrest at the mitotic decatenation checkpoint continue to proliferate due to Metnase activity. Metnase (SETMAR) is a SET-transposase fusion protein that promotes non-homologous end-joining repair even in the presence of Topo IIα inhibitor [111]. Jacoby et al. (2014) showed that DSBs response is abnormal in myeloblasts from t-AML patients, and this feature was associated with trisomy 8. The authors suggested that the association between abnormal DSBs and trisomy 8 in AML is related to MYC overexpression [112]. MYC is a proto-oncogene located at the long arm of chromosome 8 (8q24). MYC deregulation leads to DNA damage [113], the induction of genomic instability and telomere dysfunction [114]. The induction of DNA damage through DSBs seems to occur through direct MYC-mediated suppression of the NHEJ (Non-homologous end-joining), an important pathway that repairs DSBs in normal cells [115][116][117][115,116,117].
Trisomy 8 can be found in the blood of normal individuals [118][119][118,119]. Grove & Vassiliou (2014) proposed that it may be one of the early AML leukemogenesis events [120]. Importantly, the gain of chromosome 8 is one of the most common chromosomal abnormalities in AML. It represents 30–40% of cases alone or in association with other cytogenetic abnormalities, and it is known as the most frequent gain of chromosome in AML patients with CK [121][122][123][121,122,123].
2.4. Telomere Dysfunction
Telomeres are TTAGGG repetitive sequences directly associated with capping proteins, shelterin proteins, that protect the ends of chromosomes [124]. The linear chromosome DNA ends have a 3′ single-stranded overhang, which prevents those sites from being recognized as DSBs and activate DNA damage response pathways [125]. Telomere overhang length remains constant in healthy individuals over time [126][127][128][126,127,128]). However, in AML, Yan et al. (2013) reported that patients with abnormal karyotype presented shorter overhang length than those with normal karyotype [129]. The authors suggested the overhang length as an important prediction of poor prognosis in AML patients [129].
Telomeres become shorter at each cell division and, without telomerase, an enzyme that adds TTAGGG repetitive sequences to elongate the telomeres, cells undergo senescence [130]. The senescence occurs when telomeres become critically short, a phenomenon known as Hayflick limit, resulting in the cell cycle arrest. Nevertheless, cancer cells can bypass the telomere crisis through different mechanisms. Reactivation of telomerase is the most common mechanism to maintain telomere length, followed by the alternative mechanism of telomere lengthening (ALT) [131][132][131,132]. Telomerase reverse transcriptase (abbreviated as TERT, or hTERT in humans) is a catalytic subunit of the enzyme telomerase, which, together with the telomerase RNA component (TERC), comprises an important unit of the telomerase complex [133]. A decrease or loss of telomerase activity by mutations leads to telomere shortening, increasing the risk of CIN and, consequently, of cancer. The telomerase complex genes are frequently mutated in AML [134][135][134,135].
Swiggers et al. (2006) demonstrated that critically short telomeres in blasts AML patients lead to nCIN. The authors reported an increased rate of loss or gain of chromosome parts after telomere shortening [136]. Hartmann et al. (2005) also supported the relationship between short telomeres and CIN in AML. In both studies, telomere length and hTERT expression correlated with chromosomal abnormalities in AML patients. They found that telomere length in mononuclear cells of AML patients was significantly reduced compared to controls (peripheral blood granulocytes from healthy individuals). In addition, patients with abnormal karyotype presented shorter telomeres than those with normal karyotype. In contrast, extremely short telomeres (median of -3,7 kb compared to healthy donors) were found in AML patients showing multiple chromosomal abnormalities. Furthermore, hTERT continued to be associated with an increase in the karyotype complexity [137].
Capraro et al. (2012) have also shown that an abnormal karyotype was associated with shorter telomeres and extremely low telomerase activity in AML [138]. Interestingly, dyskeratosis congenita (DC), a disease characterized by bone marrow failure, also presents short telomeres and shows a high predisposition to AML (with approximately 200-fold for AML compared to the general population) [139]. Therefore, telomere shortening is viewed as an important feature in AML and to be related to poor prognosis [134][137][140][141][134,137,140,141]. However, Warny et al. (2019) reported data not corroborating with these previous studies. Their interesting findings point out that telomere length in the bone marrow mononuclear cells was similar in size both at the moment of diagnosis and at relapse. They also observed that telomere length increased after chemotherapy-induced remission, but no prognostic association was found [142]. Warny et al. (2019) correlated telomere maintenance with telomerase. This is an interesting observation since elevated telomerase activity and hTERT expression were reported in 87% of AML patients in another study [143]. Swiggers et al. (2006) also observed high telomerase activity, except that AML patients with short telomeres presented high telomerase activity. In this case, high expression of TRF1, a protein that is a negative regulator of telomere length, was proposed to explain the co-presence of high telomerase activity and extremely short telomeres [136][144][136,144].
Telomeres are also responsible for preventing end-to-end fusions of chromosomes, one of the major mechanisms that can trigger both nCIN and sCIN [145]. Critically short telomeres and, consequently, telomere aggregates can result in fused chromosomes with two centromeres (dicentric chromosomes) [146]. During anaphase, the dicentric chromosomes form a bridge between the bipolar spindles and the centromeres of the sister chromatids pulled in opposite directions causing their breakage. Importantly, such breaks can occur in different places of the chromosome, not necessarily between fused chromatids. The contiguous repetition of this process gives rise to the phenomenon known as breakage-fusion-bridge (BFB) cycles, which is associated with CIN [147][148][147,148].
Dicentric chromosomes (DC) are one of the major signatures of telomere dysfunction. The incidence of dicentric chromosomes varies among AML types, but in general is present in 8–15% of all AML cases [149][150][149,150]. DCs are mainly found in CK (23%), where more than one DC is usually present [59][151][59,151]. DCs play an important role in oncogenesis, as demonstrated by Gascoigne and Cheeseman (2013). The authors showed that the occurrence of a single dicentric chromosome could contribute to tumor initiation in AML [152]. Furthermore, Sarova et al. (2016) related the presence of DC to MDS progression to AML, in which the transformation was characterized by the acquisition of more complex karyotypes [151].
2.5. Complex Chromosomal Rearrangements
Complex chromosome rearrangements (CCR) have been extensively reported in AML [153][154][155][156][153,154,155,156]. Various mechanisms have been suggested to the occurrence of this phenomenon (e.g., non-homologous end joining (NHEJ), replication-based mechanisms, BBF cycles, telomere dysfunction) [157]. Some authors have been using the term chromothripsis for the event where genetic material suffers an enormous clustered chromosomal rearrangement on specific regions of one or few chromosomes in a single cell cycle [158]. Since chromothripsis is not proven to be the cause of this phenomenon, here we will describe these abnormalities just as CCRs [157]. Rausch et al. (2012) showed that in their cohort of AML TP53 mutated patients, ∼47% of cases presented CCRs. The occurrence of CCRs was associated with a poor prognosis [153]. Rücker et al. (2018) reported CCRs in 35% of AML patients with CK. In 85% of cases with CCRs presented mutated TP53 [159]. Once more, this data highlights the role of dysfunctional TP53 on CIN in AML. Hence, Fontana et al. (2018) have found an incidence of 6.6% CCRs in a large cohort of AML patients (N=395). It was also reported that AML cells with CCRs also presented signatures of CIN, such as TP53 alteration, a higher mean of copy number alteration (CNA), CK, 5q deletion, alterations in DNA repair, and cell cycle. They also observed that AML cells with CCRs had marker chromosomes with the MYC gene [155]. Furthermore, Gao et al. (2020) reported that in AML-MRC with CCRs, this phenomenon was associated with a lower number of white blood cells and platelets and a higher degree of karyotypic complexity. The most involved chromosomes in CCRs were the chromosomes 8 and 11, resulting in the amplification of MYC (8q24.2) or lysine methyltransferase 2A (KMT2A) (11q23.3) [160]. L′Abbate et al. (2018) analyzed MYC amplicons in AML. Their results provide evidence that CCRs are not related to a single catastrophic event as the chromothripsis model describes it but rather to an accumulative evolution [161]. Marker chromosomes are rearranged chromosomes whose genetic origin cannot be verified by conventional banding cytogenetics techniques [162]. In AML, Bochtler et al. (2017) reported that marker chromosomes could arise from CCRs and predict adverse prognosis [154]. Marker chromosomes were also suggested to be a risk classification factor for AML with adverse cytogenetics [163].
2.6. Epigenetic Regulation
Abnormalities in the epigenetic regulator Tet methylcytosine dioxygenase 2 (TET2) and Enhancer of zeste homolog 2 (EZH2) could induce CIN through the deregulation of histone modifications, which alters the chromatin structure and affect gene expression [164][165][166][164,165,166]. Mutations in the TET2 are among the most common mutations in AML [167][168][169][167,168,169]. EZH2 is located in 7q36.1, a chromosomal region affected by the loss of chromosome 7 (-7) or deletion of 7q, which reduces its gene expression [170][171][170,171]. The -7 and deletion of 7q are highly associated with CK and adverse prognosis [50][172][50,172]. Wang et al. (2020) reported that TET2 is hypermethylated (downregulated transcription) in 30% of AML patients. Alterations in the expression of TET2 or EZH2 also protects against apoptosis by an unknown mechanism [173]. Göllner et al. (2017) showed that EZH2 loss of function induced resistance to multiple drugs in AML [174]. Interestingly, the expression levels of the genes TET2 and EZH2 were also positively correlated to the CIN MAD2 and CDC20 genes expression levels [173]. The protein mitotic arrest deficient 2 (Mad2) and cell division cycle protein 20 homologue (CDC20) overexpression and downregulation are frequently altered in many cancers and associated with CIN. Both proteins are essential for the mitotic checkpoint, in which they act together as an APC/C inhibitor, preventing aneuploidy and, consequently, CIN [1][175][176][1,175,176]. Schvartzman et al. (2011) have shown, in a p53 mutant tumor model, that wtp53 represses mad2 and its upregulation is necessary for CIN in AML [174]. Overexpression of CDC20 is more present in aneuploid than euploid AML [177].