2. PVAT Modulates Vascular Function
The vascular wall of blood vessels is composed of three layers: tunica adventitia, tunica media, and tunica intima. The tunica intima, the inner layer, is a single layer of flattened, polygonal endothelial cells that rest on basal lamina and loose connective tissues, while the tunica media (especially in arteries) mainly consists of vascular smooth muscle cells (VSMC). The adventitia mainly contains connective tissue
[17]. PVAT can be found outside the adventitial layer surrounding most of the systemic blood vessels, including large arteries and veins, small and resistance vessels, and skeletal muscle microvessels. Other microvasculature and the cerebral vasculature are free of PVAT
[5][8][5,8].
The endothelium has long been recognized as an important regulator of vascular tone by releasing vasoactive factors that modulate VSMC contractility. Endothelium can release both vasodilating (such as endothelium-derived hyperpolarizing factors (EDHF), prostaglandin (PGI
2), and nitric oxide (NO)) and vasoconstricting factors (such as endothelin-1 (ET-1) and thromboxane A2 (TXA2)). The endothelium can control the vascular tone by balancing the release of these vasoactive molecules
[18]. It is well known that endothelium dysfunction is a major factor in the pathogenesis of many cardiovascular diseases, including hypertension and atherosclerosis
[19], as well as metabolic syndrome and diabetes
[20][21][20,21]. The importance of the endothelium in vascular function is undebatable; however, it is now known that it is not the sole significant regulator of the vascular tone.
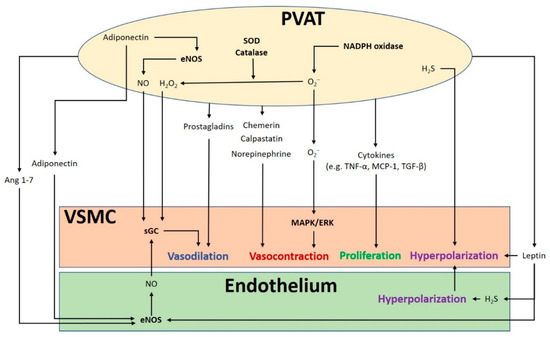
The crosstalk between PVAT and the blood vessel is vital for normal vascular function (). The modulation of the vascular function by PVAT has been demonstrated by its inhibiting effect on vascular contraction to various agonists in both rodents and humans
[3][4][3,4]. PVAT exerts anti-contractile function through the release of various PVAT-derived relaxing factors (PVRFs), previously known as the adventitium-derived relaxing factors (ADRFs)
[22]. Although it is still unclear how PVRFs exert their anti-contractile effects, a number of potential PVRFs have been suggested, including leptin and adiponectin
[23], hydrogen sulphide (H
2S)
[24], hydrogen peroxide (H
2O
2)
[19], prostaglandins
[25][26][25,26], NO
[27], and angiotensin (Ang) 1–7
[28]. Currently, it is hypothesized that PVAT modulates vascular function through two distinct mechanisms: an endothelium-dependent mechanism through a transferable PVRF and the stimulation of NO release from the endothelium, and an endothelium-independent mechanism through the generation of H
2O
2 [27]. The transferable PVRFs modulate the release of endothelial NO and the subsequent activation of potassium (K
+) channels to facilitate vascular relaxation. On the other hand, the non-transferable anticontractile property of PVAT may involve the generation of H
2O
2. A recent study suggests that the mitochondria of PVAT are important sources of O
2− and H
2O
2, which are continuously generated in response to contractile stimuli. PVAT regulates the relaxation and contraction of VSMC
[29], while ROS may act as important mediators of PVAT anticontractile effects by direct actions in the VSMC
[30].
Figure 1. The crosstalk between perivascular adipose tissue (PVAT) and the blood vessel modulates vascular function. PVAT releases vasoactive molecules. H2S is synthesized in PVAT and induces vascular smooth muscle cell (VSMC) hyperpolarization. Leptin activates eNOS and stimulates H2S production, which lead to endothelium-dependent vasodilatation. H2S released from endothelium and PVAT functions as an endothelium-derived hyperpolarizing factor (EDHF) and activates endothelial K+ channels. The resulting hyperpolarization of endothelial cells can be transmitted to VSMC. NO and H2O2 released from PVAT can elicit vasodilatation by activating soluble guanylyl cyclase (sGC). Adiponectin produced by PVAT stimulates NO production in PVAT and in endothelial cells and induces VSMC hyperpolarization. Ang 1–7 acts on endothelial cells and stimulates endothelial NO production. Vasodilating prostaglandins released from PVAT can act on VSMC and stimulate vasorelaxation. Reactive oxygen species (ROS) produced from nicotinamide adenine dinucleotide phosphate (NADPH) oxidases in PVAT can cause contractile response in VSMC via the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway. Other potential PVAT-derived contracting factors have been suggested, including chemerin, calpastatin, and norepinephrine. Antioxidant enzymes SOD and catalase in PVAT can detoxify ROS to produce H2O2, which can stimulate vasodilation. Cytokines released from PVAT, including TNF-α, MCP-1 and TGF-β can lead to VSMC proliferation. MAPK/ERK, mitogen-activated protein kinase/extracellular signal-regulated kinase; Ang, angiotensin; eNOS, endothelial nitric oxide synthase; SOD, superoxide dismutase; TNF-α, tumor necrosis factor-α; MCP-1, monocyte chemotactic protein-1; TGF-β, transforming growth factor-β; H2S, hydrogen sulphide; VSMC, vascular smooth muscle cells.
In addition to PVRF, recent studies have also demonstrated that PVAT can secrete PVAT-derived contracting factors (PVCFs), which modulate vasoconstriction
[31][32][33][31,32,33]. Studies in rats have shown that both PVRFs and PVCFs can modulate local vascular tone through endothelium-dependent or endothelium-independent effects, by as-of-yet underdetermined mechanisms
[25][34][25,34]. Currently, a number of potential PVCFs have been suggested, including chemerin
[35], calpastatin
[36], norepinephrine (NE)
[37], AngII, and ROS
[38]. Also, upon perivascular nerve stimulation, PVAT may produce superoxide (O
2−) mediated by NADPH oxidases, which enhances the arterial contractile response. This enhancement of contractile effect involves the activation of tyrosine kinase and the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway. Moreover, the contractile effects of PVAT are regulated by other antioxidant enzymes including SOD (superoxide dismutase) and catalase. Both Mn-SOD and CuZn-SOD are expressed in PVAT
[25][38][25,38]. When stimulated by NE, the expression of Mn-SOD is increased and the expression of catalase is decreased in PVAT, which induces the generation of O
2− in PVAT
[30].
It is hypothesized that mitochondria-derived ROS in PVAT modulates vascular reactivity. The uncoupling of mitochondria and the removal of H
2O
2 increase the NE-induced contraction in vessel rings surrounded with PVAT
[30]. Perivascular nerve activation by electric field stimulation (EFS) increases O
2− generation in isolated PVAT, which is attenuated by an inhibition of NADPH oxidases. Treatment with apocynin and diphenyleneiodonium chloride (DPI) to inhibit NADPH oxidases attenuates the contractile response to EFS in the vessel rings with PVAT
[38]. Also, exogenous O
2− augments the contractile response to EFS and to phenylephrine in vessel rings without PVAT
[38]. Therefore, ROS in PVAT may act as a pivotal signaling molecule in regulating the contraction of VSMC.
A recent study has shown that the anticontractile effects of PVAT can be mediated by vasoactive amines such as dopamine, NE, and serotonin, which can be uptaken and metabolized in PVAT. Monoamine oxidase A/B (MAO-A/B, which catalyzes the oxidative deamination of vasoactive amine) and semicarbazide-sensitive amine oxidase (SSAO, which catalyzes the production of H
2O
2 and ammonia) are presented in PVAT, which is responsible for the metabolism of these vasoactive amines. In rat mesenteric arteries, the inhibition of MAO and SSAO, or the inhibition of norepinephrine transporter (NET) with nisoxetine reduced PVAT’s anticontractile effect in response to NE-induced vasocontraction. Furthermore, sympathetic stimulation can trigger the release of adiponectin via β3-adrenoceptor activation in PVAT
[39]. Moreover, PVAT can also prevent noradrenaline-induced vasocontraction, by acting as a reservoir of noradrenaline and preventing it from reaching the vessel wall
[39]. These studies suggest that PVAT possesses various underdetermined mechanisms, in addition to releasing vasoactive factors, to regulate the vascular function.
3. Obesity-Linked PVAT Dysfunction
Over the past few decades, the prevalence of obesity has doubled worldwide, with a concomitant increase in associated cardiovascular diseases
[40][53]. Obesity has a variety of adverse effects on the cardiovascular system
[41][54]. Although obese patients have a higher risk of developing hypertension, cardiomyopathy, and stroke, endothelial dysfunction is not always evident in in vitro studies. In fact, the dysfunction of PVAT, but not obesity itself, is responsible for obesity-induced vascular disorders. The anticontractile effects of PVAT are attenuated in obese mice compared to lean mice
[12][42][43][12,55,56]. When PVAT is removed from the vessel, the anticontractile responses to vasodilators are not different between obese and lean mice, suggesting that obesity does not directly impair the intrinsic vascular bed reactivity but rather the PVAT function
[44][57]. Moreover, mesenteric arteries incubated with PVAT surrounding the thoracic aorta region of aorta from HFD-fed rats show reduced endothelium-dependent relaxation compared to those incubated with aortic PVAT from rats on a standard chow diet
[45][58]. Therefore, it is likely that PVAT dysfunction is related to the development of obesity-associated vascular complications.
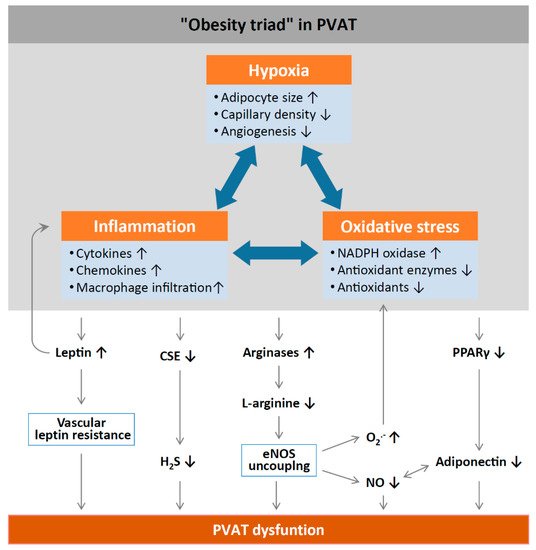
Inflammation and oxidative stress in PVAT alter the anticontractile effects under obese conditions (). HFD feeding significantly increases the mass of PVAT and the number of hypertrophic adipocytes, and results in WAT characteristics of PVAT in rodents
[45][58]. Obesity may cause inflammation in WAT-like PVAT, characterized by the infiltration of macrophage and dendritic cells with the high expression of inflammatory adipokines and cytokines, including leptin, MCP-1, tumor necrosis factor alpha (TNF-α)
[46][59], and IL-6
[47][60], while the expression of the anti-inflammatory adipokine adiponectin is reduced in obese PVAT
[48][61]. Obesity-induced PVAT inflammation also stimulates the generation of O
2− and H
2O
2 by NADPH oxidases, which promote aortic wall procontractile activity. The inactivation of O
2−, dismutation of mitochondrial-derived H
2O
2, or uncoupling of oxidative phosphorylation can reduce phenylephrine-induced vasocontraction in vessel rings surrounded by PVAT from HFD-fed mice
[12][42][12,55]. HFD-fed mice also show a reduced expression of SOD3 and glutathione levels in mesenteric PVAT
[47][60]. Also, the anticontractile effects of PVAT are impaired in mice lacking IL-18 specifically in PVAT. Moreover, these mice show increased amount of WAT-like PVAT accompanied with deformed mitochondria and decreased Mn-SOD expression
[49][62]. Obese mice lacking TNF-α receptors in PVAT have reduced H
2O
2 generation and sensitivity to phenylephrine-induced vasocontraction, suggesting that oxidative stress significantly contributes to the procontractile shift of PVAT
[42][55].
Figure 2. Mechanisms of PVAT dysfunction in diet-induced obesity. High-fat diet (HFD)-induced adipocyte hypertrophy leads to hypoxia and the production of pro-inflammatory cytokines and chemokines, the activation of NADPH oxidases, and the downregulation of antioxidant enzymes (e.g., superoxide dismutase and peroxiredoxin-1) and non-enzymatic antioxidants (e.g., glutathione). Infiltrating immune cells potentiate PVAT inflammation and oxidative stress. Chronic hyperleptinaemia leads to vascular leptin resistance (loss of leptin-induced vasodilatation) and the potentiation of PVAT inflammation. Long-term obesity decreases PVAT H
2S production by downregulating CSE expression. The upregulation of arginases leads to L-arginine deficiency and eNOS uncoupling (enhanced superoxide production and reduced NO production by eNOS). PVAT adiponectin expression is reduced in obesity, likely due to a downregulation of PPARγ. Normally, NO stimulates adiponectin secretion and adiponectin increases PVAT NO production. This positive feedback mechanism is impaired in obesity. CSE, cystathionine gamma-lyase; H
2S, hydrogen sulphide; PPARγ, peroxisome proliferator-activated receptors gamma; eNOS, endothelial nitric oxide synthase. Reproduced from Xia et al. 2017
[1] under the terms of the Creative Commons Attribution-Noncommercial License.
Aldose reductase is an enzyme of the aldoketo reductase super-family that catalyzes the conversion of glucose to sorbitol in the polyol pathway of glucose metabolism, which also depletes the antioxidant glutathione system due to the scavenging of NADPH, thereby increasing the production of ROS
[50][63]. PVAT of diabetic rats exhibits higher levels of markers of oxidative stress including augmented malonaldehyde and aldose reductase activity, which are associated with reduced antioxidant protection
[51]. In addition, increased Ang II levels may induce ROS production during obesity in systemic and related adipose tissue including PVAT
[52][64]. eNOS uncoupling and increased O
2− generation are proposed to contribute to the PVAT-induced endothelial dysfunction in obese mice
[43][56]. The impaired anticontractile effects of PVAT upon HFD feeding are not only dependent on the endothelium but are also a consequence of reduced NO bioavailability due to L-arginine deficiency and eNOS uncoupling in WAT-like PVAT
[43][53][56,65]. L-arginine supplementation and arginase inhibition could reverse obesity-induced vascular dysfunction ex vivo
[43][56]. In a human study, CuZn-SOD, peroxiredoxin-1, and adiponectin expressions are reduced in obese subjects compared to healthy subjects
[53][65]. The anti-contractility of aortic rings with normal PVAT is improved when incubated with SOD, catalase, or TNF-α, while the anticontractile effect is attenuated when aortic rings with obese PVAT are incubated with anti-TNF-α antibodies or free radical scavengers
[53][65].
PVAT inflammation could be related to hypoxia. Hypoxia stimulates the production of inflammatory cytokines and chemokines from PVAT and infiltrating macrophages
[4]. In obesity, adipocytes become hypertrophic, leading to inadequate perfusion and consequent local hypoxia. Hypoxia-inducible factor alpha (HIF-1α), a key mediator of hypoxia, is increased in the adipose tissue of obese subjects
[54][66]. HIF-1 α is responsible for the stimulation of inflammatory mediator production, such as TNF-α and IL-6, and for suppressing the expression of adiponectin
[55][67]. Incubation with TNF-α and IL-6 in PVAT leads to the attenuation of the anticontractile effects of PVAT, while the induction of hypoxia also causes inflammation and the loss of the anti-contractile function of PVAT
[4]. This hypoxia-induced PVAT dysfunction can be normalized by in vitro incubation with either anti-TNF-α antibody, anti-IL-6 antibody, or by catalase and SOD antagonists. From these observations, PVAT loses its anticontractile properties after the increase of oxidative stress during obesity. In addition, obesity related PVAT dysfunction, increased inflammation, and oxidative stress are critical features in atherogenesis. Atherosclerotic lesions are characterized by the activation of NADPH oxidases, as well as increased eNOS uncoupling and O
2− generation
[56][68]. The combination of oxidative stress and inflammation creates a vicious cycle that is further supported by a variety of metabolic and genetic risk factors leading to atherogenesis
[57][69]. Therefore, increased oxidative stress in PVAT is a critical link between obesity and vascular complications.