The correct characterisation of central nervous system (CNS) malignancies is crucial for accurate diagnosis and prognosis and also the identification of actionable genomic alterations that can guide the therapeutic strategy. Surgical biopsies are performed to characterise the tumour; however, these procedures are invasive and are not always feasible for all patients. Moreover, they only provide a static snapshot and can miss tumour heterogeneity. A liquid biopsy of the CSF can be used to obtain information about the tumour in a less invasive manner. The CSF is a source of cell-free circulating tumour DNA (ctDNA), and the analysis of this biomarker can characterise and monitor brain cancer. Recent studies have shown that ctDNA is more abundant in the CSF than plasma for CNS malignancies and that it can be sequenced to reveal tumour heterogeneity and provide diagnostic and prognostic information. Furthermore, analysis of longitudinal samples can aid patient monitoring by detecting residual disease or even tracking tumour evolution at relapse and, therefore, tailoring the therapeutic strategy.
- central nervous system malignancies
- brain cancer
- circulating tumour DNA
- cerebrospinal fluid
- liquid biopsies
1. CNS Malignant Tumor Biopsy
Central nervous system (CNS) malignancies affect both children and adults worldwide and are responsible for substantial morbidity and mortality. An epidemiological study of CNS cancer between 1990 and 2016 revealed that the age-standardised incidence rate has increased by 17.3% globally, with 330,000 incident cases and 227,000 deaths globally in 2016 [1].
CNS cancer consists of primary tumours and intracranial metastases. Most CNS tumours (>90%) occur in the brain, with the remaining located in the meninges, spinal cord and nerves. Depending on the anatomical region and the tumour type, the neurological signs and symptoms will vary and may include headaches, seizures, loss of vision, paralysis, speech disturbance, and motor deficits [2].
CNS tumours are diagnosed using neuroimaging techniques such as magnetic resonance imaging (MRI) or computed tomography; however, to obtain pathological information and molecular diagnosis, tumour biopsies are required. The treatment strategy for primary CNS tumours consists of either obtaining a biopsy or performing a surgical resection, combined, when appropriate, with postoperative radiotherapy and chemotherapy [3].
CNS tumour prognosis is diverse since there are distinct entities with different histopathological characteristics and molecular profiles. Therefore, characterising the tumour specimen is essential for accurate diagnosis and prognosis, as well as to identify potential therapeutic targets. To improve disease control, primary brain tumours or single nodule brain metastases are resected. However, obtaining tumour biopsies is not always possible due to their location, particularly when CNS tumours occur in vital regions such as the basal ganglia or the brain stem. In addition, patients with disseminated disease may not be eligible for such procedures [4][5][6].
In cases where tumour resection or obtaining a biopsy is possible, the sample obtained may not be representative of tumour heterogeneity [7][8][9], and, therefore, multiple sampling may be required to confirm the pathological diagnosis in some cases. Moreover, the analysis of the sample obtained only provides a static snapshot from the time of resection. It is important to monitor the patient’s response to treatment during the course of the disease, particularly to distinguish true disease progression from a pseudoprogression induced by treatment. Sometimes a new or enlarging area of contrast enhancement is observed, but it is not easy to assess whether it is the result of tumour growth or an inflammatory response [10]. This can be challenging when using conventional imaging techniques.
Tumours evolve over time, particularly under the selective pressure of therapy, which can result in the expansion of pre-existing resistant clones or the acquisition of
resistant alterations [7]. Thus, the genomic characteristics at relapse may differ from the genomic landscape at first occurrence. In some cases, treatment decisions at relapse are just based on the characteristics of the primary tissue obtained [11][12]. The known evolution of tumours and the absence of longitudinal tumour sampling may, therefore, lead to imprecise diagnosis and clinical management.
For these reasons, there is an urgent need to develop less invasive methods to identify and validate tumour biomarkers that provide real-time information to aid in diagnosing and monitoring CNS malignancies. Overall, this will help to adjust the therapeutic strategy and guide treatment decisions based on the current tumour profile and its burden.
An alternative to a tumour biopsy is a liquid biopsy (
). Liquid biopsies are emerging as noninvasive tools that can provide longitudinal information about the tumour genomic landscape and facilitate patient monitoring. It consists of the analysis of biomarkers, including circulating tumour cells, exosomes and circulating tumour nucleic acids that are present in bodily fluids such as blood, cerebrospinal fluid (CSF), urine and saliva [13][14].
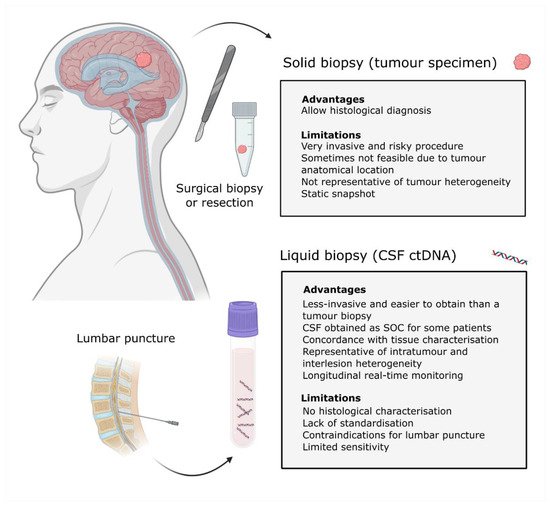
Solid vs. liquid biopsies. Schematic representation of the tumour biopsy and CSF samples obtained from a patient with a CNS malignancy. Advantages and limitations for each methodology are indicated. Definition of acronyms: standard of care (SOC), cerebrospinal fluid (CSF), circulating tumour DNA (cfDNA) and central nervous system (CNS).
2. Circulating Cell-Free DNA and Circulating Tumour DNA
Cells release DNA that then circulate in bodily fluids. The fraction of cell-free DNA (cfDNA) that is shed by cancer cells, presumably undergoing apoptosis or necrosis, is known as ctDNA and carries genomic alterations that can be detected using PCR-based or next-generation sequencing (NGS)-based methods [14][15][16][17].
Increased concentrations of cfDNA have been detected in pathological conditions like trauma, infection and cancer, or even other physiological conditions like exercise [18].
For patients with an extracranial disease, plasma ctDNA has been detected across different cancer types. However, blood may not be a suitable source from patients with CNS malignancies since ctDNA levels were infrequently detected in plasma [19][20]. ctDNA was detectable in the plasma of >75% of patients with advanced cancers, such as bladder, colorectal, gastroesophageal, ovarian, pancreatic, breast, melanoma, hepatocellular, and head and neck cancers, in contrast with <10% of glioma patients (2/27) [19].
The proportion of ctDNA in the blood is small and varies depending on tumour characteristics, including type, grade and burden [16][19]. In contrast, the total amount of ctDNA in CSF is increased, making it an ideal biofluid to characterise and monitor CNS cancer [20][21]. Interestingly, the levels of ctDNA in CSF may be influenced by tumour burden, tumour progression and anatomical location of the tumour, with regard to the proximity to CSF reservoirs [21][22][23].
3. Cerebrospinal Fluid as a Source of ctDNA
CSF is a clear bodily fluid secreted by the choroid plexus that is present in the subarachnoid space of the brain, the spinal cord and the central canal [24].
CSF is in direct contact with the brain parenchyma, and several studies have shown that CSF is a reliable source of cell-free ctDNA, providing advantages over plasma or serum for the analysis of CNS tumours [20][21].
Several studies have reported the ability to detect ctDNA in CSF of patients with CNS malignancies. Gene mutations and molecular alterations have been detected in the CSF DNA of CNS cancer patients [25][26][27][28][29][30], followed by genomic landscape characterisation of CSF ctDNA with the development of high throughput sequencing technologies [20][21][22][23][31][32][33][34][35][36].
CSF samples can be accessed through a lumbar puncture or obtained from the ventricles under certain circumstances. Patients with posterior fossa tumours tend to present with hydrocephalus, a condition in which the CSF accumulates within the cerebral ventricles and/or subarachnoid spaces [37][38][39]. A lumbar puncture is contraindicated in these patients, given the risk of brain herniation; therefore, CSF is obtained from the ventricles during procedures that are performed to alleviate intracranial pressure and drain the excess of CSF [38][40][41][42].
As part of the CNS staging criteria for certain brain tumours, a diagnostic lumbar puncture is routinely performed as standard of care for CSF cytology evaluation of CNS lymphoma, CNS metastasis, and medulloblastoma [43][44][45][46]. In these cases, the CSF samples collected as standard of care can be further used to characterise the ctDNA and provide information about the tumour [20][34][47].
4. Clinical Applications of the CSF ctDNA for CNS Malignancies
CNS tumours are a heterogeneous group of malignancies [48]. In most cases, tumour resection is required to reduce tumour burden and mass effect [49]. However, liquid biopsies can be used to complement histopathological diagnosis and are essential for those patients with inoperable tumours.
The monitoring of CNS malignancies is currently performed by imaging techniques; however, these are not sensitive for microscopic disease [50]. A complementary liquid biopsy of CSF could, therefore, be performed to aid clinical assessment by determining the response to treatment, differentiating pseudoprogression from true progression and tracking levels of residual disease. In addition, the genomic characterisation of CSF ctDNA can facilitate the identification of actionable genomic alterations that confer sensitivity or resistance to clinically available drugs and the detection of mechanisms of resistance at relapse.
The distinct clinical applications of the analysis of CSF ctDNA are discussed below for patients with distinct types of CNS cancer.
4.1. Diffuse Gliomas
4.1.1. A Diagnostic and Prognostic Tool
Among diffuse gliomas, glioblastoma (GBM) is the most common malignant brain tumour in adults, with a 2-year survival of 18% and 5-year overall survival (OS) of 4% [51]. Providing an accurate molecular profile for diagnosis and prognosis is essential and can be achieved with a CSF liquid biopsy. The analysis of the mutational status of
,
,
,
,
,
and
in CSF ctDNA facilitates the molecular diagnosis of diffuse gliomas and provides prognostic information in a relatively noninvasive manner [22]. In addition,
promoter mutations have been detected in the CSF ctDNA of GBM patients, and shorter OS of patients with high variant allele frequency (VAF) has been observed. The results from this pilot study suggested that VAF levels of the
promoter mutation could be a predictor of poor survival [52]. In more recent studies, CSF was obtained from lumbar punctures in glioma patients, and ctDNA was detected and was associated with disease burden, tumour progression, and adverse outcomes [20][23]. Moreover, most patients with detectable ctDNA had a negative cytopathologic analysis, and ctDNA was not detected in plasma [20][23].
Diffuse midline glioma (DMG) is a tumour entity characterised by a K27M mutation in either
or
; it is usually located in the brain stem, thalamus and spinal cord [48]. Within H3 K27M-mutant DMG, diffuse intrinsic pontine glioma (DIPG) is a rapidly growing tumour in the brain stem that typically arises in young children and is associated with poor survival [53]. The anatomical location of these tumours, the brainstem, makes them difficult and dangerous to biopsy. Importantly, H3 K27M mutations can be detected in the CSF ctDNA, facilitating diagnosis and opening the possibility of avoiding diagnostic surgical biopsies [22][54][55].
The molecular characterisation and understanding of DIPG biology have been improved from specimens obtained from rare diagnostic biopsies and postmortem tissue donations [56][57][58][59]. Indeed, the lack of surgical specimens can be overcome by the analysis of CSF ctDNA to aid in the management of patients with DIPG and contribute to the molecular study of this disease to accelerate research. An NGS panel of 68 genes commonly mutated in brainstem tumours was used to study a cohort of 57 patients with brainstem tumours, including 23 patients with DIPG. Mutations were detected in the CSF ctDNA of 82.5% of patients, and the presence of
mutations was correlated with poor OS while the
mutation predicted better OS [54]. Moreover, longitudinal analysis of CSF samples offers the possibility of monitoring and allows the tumour evolution of this dismal disease to be studied.
4.1.2. Monitoring and Therapeutic Strategies
The number of actionable genomic alterations for patients with primary brain tumours is limited. The most relevant biomarker for glioma is
promoter methylation status.
promoter methylation causes the loss of
expression, and since it is involved in DNA repair by reversing DNA alkylation,
promoter methylation renders cells more susceptible to temozolomide and is associated with longer survival [60][61][62][63].
promoter methylation was detected using methylation-specific PCR from genomic DNA extracted from the CSF of glioma patients, with higher sensitivity than from serum [64].
A potential biomarker for GBM is epidermal growth factor receptor (
).
amplification and EGFRvIII mutation have been detected in RNA within extracellular vesicles circulating in CSF [65]. This could be of high interest as a biomarker to predict response to future EGFRvIII-targeted therapies in GBMs.
4.2. Brain and Leptomeningeal Metastases
4.2.1. CSF ctDNA Facilitates Diagnosis and Allows Tumour Genomic Characterisation
About 20–40% of patients with advanced-stage cancers of the lung, breast and melanoma develop brain metastasis, and approximately 5–8% of these patients are diagnosed with leptomeningeal metastasis. These are devastating diseases that carry a poor prognosis and are often resistant to treatment [66][67][68].
In addition to brain metastasis present in the brain parenchyma, malignant cells can seed the leptomeninges, causing leptomeningeal metastasis [67][69][70]. The diagnosis of leptomeningeal metastasis is based on clinical symptoms, MRI scans, and cytology analysis of CSF [71]. However, up to 20% of patients with positive clinical and radiographic signs presented false-negative CSF cytology [72][73]. Several studies have shown that ctDNA can be detected in the CSF of patients with negative cytology analysis [20][27][32][74]. Cytology has limited sensitivity, and the analysis of ctDNA can complement the diagnosis of leptomeningeal metastases.
Brain metastasis can present different genomic alterations compared to their primary extracranial tumour [12]. Several studies have shown that the analysis of CSF ctDNA enables the characterisation of the genomic complexity of CNS metastases, including intratumour heterogeneity, revealed with the identification of trunk and private genomic alterations. Moreover, the genomic landscape of CNS metastasis, including the brain lesion’s private alterations, was better represented from the ctDNA in the CSF than plasma [20][32][75]. Analysis of CSF ctDNA from a cohort of 26 patients with leptomeningeal metastases from non-small cell lung cancer (NSCLC) revealed their unique genetic profiles, including mutations in several driver genes, copy number variations (CNVs) in
,
,
,
, and
, and loss of heterozygosity in
[76].
4.2.2. Patient Monitoring and Identification of Therapeutic Targets
There are several targeted therapies for brain metastases [77][78][79][80][81][82][83][84][85][86]. For EGFR-mutated brain metastases from NSCLC, first-, second- and third- generation EGFR tyrosine kinase inhibitors (TKIs) are available [87][88][89][90]. In addition, for NSCLC with ALK gene rearrangement, CNS penetration and therapeutic potential were exhibited by second-generation ALK inhibitors [84][85][86]. There are also targeted therapies for patients diagnosed with HER2+ breast cancer and melanoma patients, including BRAF and MEK inhibitors [82][83][91][92]. For the treatment of leptomeningeal metastases, targeted therapies for the aforementioned actionable genomic alterations may also be effective [93].
The availability of targeted therapies highlights the importance of the identification of actionable genomic alterations or resistance mutations in genes, including
,
,
and
, which have been detected from CSF ctDNA in several studies [20][21][31][32][94][95][96]. For example, EGFR–TKI resistance mutation
T790M has been detected in the CSF ctDNA of lung cancer patients [94][97].
A study of 21 patients with brain metastasis from NSCLC compared the NGS results obtained from different samples to reveal the mutation pattern of driver genes for each patient. Mutations were detected in 95.2%, 66.7% and 39% of patients from CSF ctDNA, plasma ctDNA, and plasma circulating tumour cells, respectively. The most mutated gene was
followed by
,
,
,
,
,
,
and
(all >15%). For
mutations, the detection rate was 57.1% (12/21) from CSF ctDNA, which, interestingly, was higher for patients with leptomeningeal (81.8%; 9/11) compared with brain parenchymal (30%; 3/10) metastases [98].
The analysis of CSF ctDNA can also contribute to monitoring response to treatment. Metastasis in the CNS developed in a patient with HER2+ breast cancer. Analysis of baseline CSF ctDNA revealed mutations in
and
and amplification in
and c
. Following treatment with T-DM1, extracranial disease control was achieved, and marker levels in plasma decreased. However, the levels increased in CSF ctDNA, consistent with poor treatment benefit to the CNS [99].
Altogether, these results indicate that CSF is a more suitable fluid than plasma to reveal the mutational profile of CNS metastases and can aid in diagnosis, tailored treatment selection and monitoring.
4.3. CNS Lymphoma
Malignant B-cells can infiltrate the CNS and are associated with poor prognosis, particularly at relapse [100]. Primary CNS lymphoma is defined by the absence of systemic disease in contrast to secondary CNS lymphoma that presents infiltration into the CNS with previous or concomitant systemic lymphoma [101][102].
ctDNA has only been detected in the plasma of a minority of patients with restricted CNS lymphoma [103][104]. In contrast, several studies of patients with CNS lymphomas detected ctDNA in CSF [47][105][106][107][108][109].
Diagnosis and monitoring of CNS lymphoma are challenging, given the difficulties of tumour biopsies and the lack of sensitivity of CSF standard analysis (cytology and flow cytometry) and neuroimaging. The detection of the
L265P mutation strongly suggests the diagnosis of primary CNS lymphoma, and this mutation has been detected in the CSF ctDNA of patients with CNS lymphoma [47][106][108][109], showing that the analysis of CSF ctDNA could complement the diagnosis.
An NGS-based analysis of the CSF cfDNA of 8 patients with CNS lymphoma, at recurrence, detected tumour-derived genetic alterations and showed that the clearance of ctDNA from CSF was associated with sustained tumour responses [105].
The comparison of CSF ctDNA with plasma ctDNA and CSF standard analysis (cytology and flow cytometry) revealed that the analysis of CSF ctDNA better detected CNS disease in patients with B-cell lymphoma [47]. Moreover, longitudinal analysis of CSF ctDNA levels allowed the monitoring of response to treatment, the detection of residual disease and predicted relapse. The dynamic changes observed in CSF ctDNA recapitulated the evolution of the disease for patients with CNS lymphoma [47].
4.4. Medulloblastoma
CNS tumours are the leading cause of cancer-related mortality in children and adolescents due to the aggressiveness of certain subtypes, including medulloblastoma and high-grade gliomas such as DMG [110][111].
Medulloblastoma (MB), an embryonal tumour of the CNS, is the most aggressive brain tumour in childhood that can also occur in adults, although this is less common [112]. MB is a complex and evolving heterogeneous disease that can be divided into four molecular consensus subgroups (WNT, SHH, Group 3 and Group 4), with further subtypes identified [113][114][115][116][117]. The lack of sufficient sample, intratumour heterogeneity or the presence of disseminated disease make diagnosis and monitoring difficult [8][118][119][120][121]. However, hydrocephalus is common amongst these patients, and CSF samples can be obtained prior to tumour surgical resection or biopsy [40][42]. In addition, CSF samples are routinely collected through a lumbar puncture for cytology analysis to assess metastatic dissemination according to Chang’s M-staging system, in combination with brain and spinal MRIs [45].
The study of paediatric patients with MB showed that ctDNA was more abundant in CSF (76.9%, 10/13 patients) than plasma (1/13 patients) for patients with negative CSF cytology results. Moreover, exome sequencing of CSF ctDNA recapitulated the tumour mutational burden and the genomic alterations, including MB common mutations (
,
), CNVs (
and
amplification) and arm-level chromosomal aberrations (chromosome 17p loss), providing diagnostic and prognostic information [34]. Longitudinal CSF samples were also collected, and ctDNA analysis detected residual disease, identified intratumour and interlesion heterogeneity, and revealed a genomic transformation of the tumour at relapse [34]. More recently, another study reported the detection of ctDNA in CSF; however, shared genetic mutations between CSF and the tumour specimen were only identified in 22% (2/9) of patients. The authors suggested that this could be explained by the time-interval differences between tumour and CSF collection [122].
MB also presents abnormal DNA methylation changes, with distinct epigenetic signatures identified across MB subtypes that can be altered during tumour progression and treatment [114][123]. The epigenetic analysis of CSF ctDNA from 4 MB patients (3 with matching tumour samples) was attained. A positive correlation of tumour and CSF samples was identified, suggesting that CSF ctDNA could be used to monitor changes in MB tumour DNA methylomes and hydroxymethylomes. In addition, DNA methylation markers of diagnostic and prognostic value could be detected in the CSF ctDNA [124]. In summary, CSF ctDNA analysis could facilitate the clinical management of paediatric patients with MB.
Further information regarding the clinical applications of the CSF ctDNA for CNS malignancies, the challenges/limitations that we need to overcome and the future insights for its implementation in the clinical setting, can be found in: Escudero, L.; Martínez-Ricarte, F.; Seoane, J. ctDNA-Based Liquid Biopsy of Cerebrospinal Fluid in Brain Cancer. Cancers 2021, 13, 1989.
References
- Patel, A.P.; Fisher, J.L.; Nichols, E.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; Fitzmaurice, C. Global, regional, and national burden of brain and other CNS cancer, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 376–393.
- Alentorn, A.; Hoang-Xuan, K.; Mikkelsen, T. Presenting signs and symptoms in brain tumors. Handb. Clin. Neurol. 2016, 134, 19–26.
- Preusser, M.; Marosi, C. Neuro-oncology in 2016: Advances in brain tumour classification and therapy. Nat. Rev. Neurol. 2017, 13, 71–72.
- Owen, S.; Souhami, L. The management of brain metastases in non-small cell lung cancer. Front. Oncol. 2014, 4, 248.
- Ferguson, S.D.; Wagner, K.M.; Prabhu, S.S.; McAleer, M.F.; McCutcheon, I.E.; Sawaya, R. Neurosurgical management of brain metastases. Clin. Exp. Metastasis 2017, 34, 377–389.
- Wijdicks, E.F.M. Historical awareness of the brainstem: From a subsidiary structure to a vital center. Neurology 2020, 95, 484–488.
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94.
- Morrissy, A.S.; Cavalli, F.M.G.; Remke, M.; Ramaswamy, V.; Shih, D.J.H.; Holgado, B.L.; Farooq, H.; Donovan, L.K.; Garzia, L.; Agnihotri, S.; et al. Spatial heterogeneity in medulloblastoma. Nat. Genet. 2017, 49, 780–788.
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014.
- Thust, S.C.; van den Bent, M.J.; Smits, M. Pseudoprogression of brain tumors. J. Magn. Reason. Imaging 2018, 48, 571–589.
- Johnson, B.E.; Mazor, T.; Hong, C.; Barnes, M.; Aihara, K.; McLean, C.Y.; Fouse, S.D.; Yamamoto, S.; Ueda, H.; Tatsuno, K.; et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 2014, 343, 189–193.
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177.
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: Genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586.
- Siravegna, G.; Mussolin, B.; Venesio, T.; Marsoni, S.; Seoane, J.; Dive, C.; Papadopoulos, N.; Kopetz, S.; Corcoran, R.B.; Siu, L.L.; et al. How liquid biopsies can change clinical practice in oncology. Ann. Oncol. 2019, 30, 1580–1590.
- Schwarzenbach, H.; Hoon, D.S.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437.
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238.
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2019, 20, 71–88.
- Crigna, A.T.; Samec, M.; Koklesova, L.; Liskova, A.; Giordano, F.A.; Kubatka, P.; Golubnitschaja, O. Cell-free nucleic acid patterns in disease prediction and monitoring-hype or hope? EPMA J. 2020, 11, 1–25.
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early-and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra224.
- De Mattos-Arruda, L.; Mayor, R.; Ng, C.K.Y.; Weigelt, B.; Martínez-Ricarte, F.; Torrejon, D.; Oliveira, M.; Arias, A.; Raventos, C.; Tang, J.; et al. Cerebrospinal fluid-derived circulating tumour DNA better represents the genomic alterations of brain tumours than plasma. Nat. Commun. 2015, 6, 8839.
- Wang, Y.; Springer, S.; Zhang, M.; McMahon, K.W.; Kinde, I.; Dobbyn, L.; Ptak, J.; Brem, H.; Chaichana, K.; Gallia, G.L.; et al. Detection of tumor-derived DNA in cerebrospinal fluid of patients with primary tumors of the brain and spinal cord. Proc. Natl. Acad. Sci. USA 2015, 112, 9704–9709.
- Martínez-Ricarte, F.; Mayor, R.; Martínez-Sáez, E.; Rubio-Pérez, C.; Pineda, E.; Cordero, E.; Cicuéndez, M.; Poca, M.A.; López-Bigas, N.; Ramon, Y.C.S.; et al. Molecular Diagnosis of Diffuse Gliomas through Sequencing of Cell-Free Circulating Tumor DNA from Cerebrospinal Fluid. Clin. Cancer Res. 2018, 24, 2812–2819.
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 2019, 565, 654–658.
- Ghersi-Egea, J.F.; Strazielle, N.; Catala, M.; Silva-Vargas, V.; Doetsch, F.; Engelhardt, B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol. 2018, 135, 337–361.
- Schmitt-Gräff, A.; Hummel, M.; Anagnostopoulos, I.; Stoltenburg, G.; Stein, H. Primary brain lymphoma in acquired immunodeficiency syndrome. Immunophenotype and molecular pathologic characterization in stereotactic biopsy, autopsy and cerebrospinal fluid cytology. Pathologe 1995, 16, 75–80.
- Rhodes, C.H.; Honsinger, C.; Sorenson, G.D. PCR-detection of tumor-derived p53 DNA in cerebrospinal fluid. Am. J. Clin. Pathol. 1995, 103, 404–408.
- Swinkels, D.W.; de Kok, J.B.; Hanselaar, A.; Lamers, K.; Boerman, R.H. Early detection of leptomeningeal metastasis by PCR examination of tumor-derived K-ras DNA in cerebrospinal fluid. Clin. Chem. 2000, 46, 132–133.
- Shi, W.; Lv, C.; Qi, J.; Zhao, W.; Wu, X.; Jing, R.; Wu, X.; Ju, S.; Chen, J. Prognostic value of free DNA quantification in serum and cerebrospinal fluid in glioma patients. J. Mol. Neurosci. 2012, 46, 470–475.
- Yang, H.; Cai, L.; Zhang, Y.; Tan, H.; Deng, Q.; Zhao, M.; Xu, X. Sensitive detection of EGFR mutations in cerebrospinal fluid from lung adenocarcinoma patients with brain metastases. J. Mol. Diagn. 2014, 16, 558–563.
- Touat, M.; Duran-Peña, A.; Alentorn, A.; Lacroix, L.; Massard, C.; Idbaih, A. Emerging circulating biomarkers in glioblastoma: Promises and challenges. Expert Rev. Mol. Diagn. 2015, 15, 1311–1323.
- Pan, W.; Gu, W.; Nagpal, S.; Gephart, M.H.; Quake, S.R. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem. 2015, 61, 514–522.
- Pentsova, E.I.; Shah, R.H.; Tang, J.; Boire, A.; You, D.; Briggs, S.; Omuro, A.; Lin, X.; Fleisher, M.; Grommes, C.; et al. Evaluating Cancer of the Central Nervous System Through Next-Generation Sequencing of Cerebrospinal Fluid. J. Clin. Oncol. 2016, 34, 2404–2415.
- Mouliere, F.; Mair, R.; Chandrananda, D.; Marass, F.; Smith, C.G.; Su, J.; Morris, J.; Watts, C.; Brindle, K.M.; Rosenfeld, N. Detection of cell-free DNA fragmentation and copy number alterations in cerebrospinal fluid from glioma patients. EMBO Mol. Med. 2018, 10.
- Escudero, L.; Llort, A.; Arias, A.; Diaz-Navarro, A.; Martínez-Ricarte, F.; Rubio-Perez, C.; Mayor, R.; Caratù, G.; Martínez-Sáez, E.; Vázquez-Méndez, É.; et al. Circulating tumour DNA from the cerebrospinal fluid allows the characterisation and monitoring of medulloblastoma. Nat. Commun. 2020, 11, 5376.
- Seoane, J.; De Mattos-Arruda, L.; Le Rhun, E.; Bardelli, A.; Weller, M. Cerebrospinal fluid cell-free tumour DNA as a liquid biopsy for primary brain tumours and central nervous system metastases. Ann. Oncol. 2019, 30, 211–218.
- Le Rhun, E.; Seoane, J.; Salzet, M.; Soffietti, R.; Weller, M. Liquid biopsies for diagnosing and monitoring primary tumors of the central nervous system. Cancer Lett. 2020, 480, 24–28.
- Nguyen, T.T.; Smith, M.V.; Rodziewicz, G.S.; Lemke, S.M. Hydrocephalus caused by metastatic brain lesions: Treatment by third ventriculostomy. J. Neurol. Neurosurg. Psychiatry 1999, 67, 552–553.
- Marx, S.; Reinfelder, M.; Matthes, M.; Schroeder, H.W.S.; Baldauf, J. Frequency and treatment of hydrocephalus prior to and after posterior fossa tumor surgery in adult patients. Acta Neurochir. 2018, 160, 1063–1071.
- Won, S.Y.; Dubinski, D.; Behmanesh, B.; Bernstock, J.D.; Seifert, V.; Konczalla, J.; Tritt, S.; Senft, C.; Gessler, F. Management of hydrocephalus after resection of posterior fossa lesions in pediatric and adult patients-predictors for development of hydrocephalus. Neurosurg. Rev. 2020, 43, 1143–1150.
- Sainte-Rose, C.; Cinalli, G.; Roux, F.E.; Maixner, R.; Chumas, P.D.; Mansour, M.; Carpentier, A.; Bourgeois, M.; Zerah, M.; Pierre-Kahn, A.; et al. Management of hydrocephalus in pediatric patients with posterior fossa tumors: The role of endoscopic third ventriculostomy. J. Neurosurg. 2001, 95, 791–797.
- Engelborghs, S.; Niemantsverdriet, E.; Struyfs, H.; Blennow, K.; Brouns, R.; Comabella, M.; Dujmovic, I.; van der Flier, W.; Frölich, L.; Galimberti, D.; et al. Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimer Dement. 2017, 8, 111–126.
- Lee, M.; Wisoff, J.H.; Abbott, R.; Freed, D.; Epstein, F.J. Management of hydrocephalus in children with medulloblastoma: Prognostic factors for shunting. Pediatr. Neurosurg. 1994, 20, 240–247.
- Balhuizen, J.C.; Bots, G.T.; Schaberg, A.; Bosman, F.T. Value of cerebrospinal fluid cytology for the diagnosis of malignancies in the central nervous system. J. Neurosurg. 1978, 48, 747–753.
- Cohen, N.R.; Phipps, K.; Harding, B.; Jacques, T.S. Is CSF cytology a useful diagnostic procedure in staging paediatric CNS tumours? Cytopathology 2009, 20, 256–260.
- Chang, C.H.; Housepian, E.M.; Herbert, C., Jr. An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 1969, 93, 1351–1359.
- Rahimi, J.; Woehrer, A. Overview of cerebrospinal fluid cytology. Handb. Clin. Neurol. 2017, 145, 563–571.
- Bobillo, S.; Crespo, M.; Escudero, L.; Mayor, R.; Raheja, P.; Carpio, C.; Rubio-Perez, C.; Tazón-Vega, B.; Palacio, C.; Carabia, J.; et al. Cell free circulating tumor DNA in cerebrospinal fluid detects and monitors central nervous system involvement of B-cell lymphomas. Haematologica 2021, 106, 513–521.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820.
- Rees, J.H. Diagnosis and treatment in neuro-oncology: An oncological perspective. Br. J. Radiol. 2011, 84, S82–S89.
- Aquino, D.; Gioppo, A.; Finocchiaro, G.; Bruzzone, M.G.; Cuccarini, V. MRI in Glioma Immunotherapy: Evidence, Pitfalls, and Perspectives. J. Immunol. Res. 2017, 2017, 5813951.
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-term (≥ 2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 11622.
- Juratli, T.A.; Stasik, S.; Zolal, A.; Schuster, C.; Richter, S.; Daubner, D.; Juratli, M.A.; Thowe, R.; Hennig, S.; Makina, M.; et al. TERT Promoter Mutation Detection in Cell-Free Tumor-Derived DNA in Patients with IDH Wild-Type Glioblastomas: A Pilot Prospective Study. Clin. Cancer Res. 2018, 24, 5282–5291.
- Hoffman, L.M.; Veldhuijzen van Zanten, S.E.M.; Colditz, N.; Baugh, J.; Chaney, B.; Hoffmann, M.; Lane, A.; Fuller, C.; Miles, L.; Hawkins, C.; et al. Clinical, Radiologic, Pathologic, and Molecular Characteristics of Long-Term Survivors of Diffuse Intrinsic Pontine Glioma (DIPG): A Collaborative Report from the International and European Society for Pediatric Oncology DIPG Registries. J. Clin. Oncol. 2018, 36, 1963–1972.
- Pan, C.; Diplas, B.H.; Chen, X.; Wu, Y.; Xiao, X.; Jiang, L.; Geng, Y.; Xu, C.; Sun, Y.; Zhang, P.; et al. Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta Neuropathol. 2019, 137, 297–306.
- Azad, T.D.; Jin, M.C.; Bernhardt, L.J.; Bettegowda, C. Liquid biopsy for pediatric diffuse midline glioma: A review of circulating tumor DNA and cerebrospinal fluid tumor DNA. Neurosurg. Focus 2020, 48, E9.
- Kambhampati, M.; Perez, J.P.; Yadavilli, S.; Saratsis, A.M.; Hill, A.D.; Ho, C.Y.; Panditharatna, E.; Markel, M.; Packer, R.J.; Nazarian, J. A standardized autopsy procurement allows for the comprehensive study of DIPG biology. Oncotarget 2015, 6, 12740–12747.
- Kambhampati, M.; Panditharatna, E.; Yadavilli, S.; Saoud, K.; Lee, S.; Eze, A.; Almira-Suarez, M.I.; Hancock, L.; Bonner, E.R.; Gittens, J.; et al. Harmonization of postmortem donations for pediatric brain tumors and molecular characterization of diffuse midline gliomas. Sci. Rep. 2020, 10, 10954.
- Baugh, J.; Bartels, U.; Leach, J.; Jones, B.; Chaney, B.; Warren, K.E.; Kirkendall, J.; Doughman, R.; Hawkins, C.; Miles, L.; et al. The international diffuse intrinsic pontine glioma registry: An infrastructure to accelerate collaborative research for an orphan disease. J. Neurooncol. 2017, 132, 323–331.
- Veldhuijzen van Zanten, S.E.M.; Lane, A.; Heymans, M.W.; Baugh, J.; Chaney, B.; Hoffman, L.M.; Doughman, R.; Jansen, M.H.A.; Sanchez, E.; Vandertop, W.P.; et al. External validation of the diffuse intrinsic pontine glioma survival prediction model: A collaborative report from the International DIPG Registry and the SIOPE DIPG Registry. J. Neurooncol. 2017, 134, 231–240.
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003.
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 2008, 26, 4189–4199.
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and Therapeutic Biomarkers in Glioblastoma: Current Status and Future Perspectives. Biomed. Res. Int. 2017, 2017, 8013575.
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329.
- Wang, Z.; Jiang, W.; Wang, Y.; Guo, Y.; Cong, Z.; Du, F.; Song, B. MGMT promoter methylation in serum and cerebrospinal fluid as a tumor-specific biomarker of glioma. Biomed. Rep. 2015, 3, 543–548.
- Figueroa, J.M.; Skog, J.; Akers, J.; Li, H.; Komotar, R.; Jensen, R.; Ringel, F.; Yang, I.; Kalkanis, S.; Thompson, R.; et al. Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro Oncol. 2017, 19, 1494–1502.
- Tabouret, E.; Chinot, O.; Metellus, P.; Tallet, A.; Viens, P.; Gonçalves, A. Recent trends in epidemiology of brain metastases: An overview. Anticancer Res. 2012, 32, 4655–4662.
- Le Rhun, E.; Weller, M.; Brandsma, D.; Van den Bent, M.; de Azambuja, E.; Henriksson, R.; Boulanger, T.; Peters, S.; Watts, C.; Wick, W.; et al. EANO-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up of patients with leptomeningeal metastasis from solid tumours. Ann. Oncol. 2017, 28, iv84–iv99.
- Beauchesne, P. Intrathecal chemotherapy for treatment of leptomeningeal dissemination of metastatic tumours. Lancet Oncol. 2010, 11, 871–879.
- Remon, J.; Le Rhun, E.; Besse, B. Leptomeningeal carcinomatosis in non-small cell lung cancer patients: A continuing challenge in the personalized treatment era. Cancer Treat. Rev. 2017, 53, 128–137.
- Dudani, S.; Mazzarello, S.; Hilton, J.; Hutton, B.; Vandermeer, L.; Fernandes, R.; Ibrahim, M.F.; Smith, S.; Majeed, H.; Al-Baimani, K.; et al. Optimal Management of Leptomeningeal Carcinomatosis in Breast Cancer Patients-A Systematic Review. Clin. Breast Cancer 2016, 16, 456–470.
- Pan, Z.; Yang, G.; He, H.; Yuan, T.; Wang, Y.; Li, Y.; Shi, W.; Gao, P.; Dong, L.; Zhao, G. Leptomeningeal metastasis from solid tumors: Clinical features and its diagnostic implication. Sci. Rep. 2018, 8, 10445.
- Glantz, M.J.; Cole, B.F.; Glantz, L.K.; Cobb, J.; Mills, P.; Lekos, A.; Walters, B.C.; Recht, L.D. Cerebrospinal fluid cytology in patients with cancer: Minimizing false-negative results. Cancer 1998, 82, 733–739.
- Clarke, J.L.; Perez, H.R.; Jacks, L.M.; Panageas, K.S.; Deangelis, L.M. Leptomeningeal metastases in the MRI era. Neurology 2010, 74, 1449–1454.
- Zhao, Y.; He, J.Y.; Zou, Y.L.; Guo, X.S.; Cui, J.Z.; Guo, L.; Bu, H. Evaluating the cerebrospinal fluid ctDNA detection by next-generation sequencing in the diagnosis of meningeal Carcinomatosis. BMC Neurol. 2019, 19, 331.
- Ge, M.; Zhan, Q.; Zhang, Z.; Ji, X.; Zhou, X.; Huang, R.; Liang, X. Different next-generation sequencing pipelines based detection of tumor DNA in cerebrospinal fluid of lung adenocarcinoma cancer patients with leptomeningeal metastases. BMC Cancer 2019, 19, 143.
- Li, Y.S.; Jiang, B.Y.; Yang, J.J.; Zhang, X.C.; Zhang, Z.; Ye, J.Y.; Zhong, W.Z.; Tu, H.Y.; Chen, H.J.; Wang, Z.; et al. Unique genetic profiles from cerebrospinal fluid cell-free DNA in leptomeningeal metastases of EGFR-mutant non-small-cell lung cancer: A new medium of liquid biopsy. Ann. Oncol. 2018, 29, 945–952.
- Rochet, N.M.; Kottschade, L.A.; Markovic, S.N. Vemurafenib for melanoma metastases to the brain. N. Engl. J. Med. 2011, 365, 2439–2441.
- Welsh, J.W.; Komaki, R.; Amini, A.; Munsell, M.F.; Unger, W.; Allen, P.K.; Chang, J.Y.; Wefel, J.S.; McGovern, S.L.; Garland, L.L.; et al. Phase II trial of erlotinib plus concurrent whole-brain radiation therapy for patients with brain metastases from non-small-cell lung cancer. J. Clin. Oncol. 2013, 31, 895–902.
- Seoane, J.; De Mattos-Arruda, L. Brain metastasis: New opportunities to tackle therapeutic resistance. Mol. Oncol. 2014, 8, 1120–1131.
- Long, G.V.; Trefzer, U.; Davies, M.A.; Kefford, R.F.; Ascierto, P.A.; Chapman, P.B.; Puzanov, I.; Hauschild, A.; Robert, C.; Algazi, A.; et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): A multicentre, open-label, phase 2 trial. Lancet Oncol. 2012, 13, 1087–1095.
- Margolin, K.; Ernstoff, M.S.; Hamid, O.; Lawrence, D.; McDermott, D.; Puzanov, I.; Wolchok, J.D.; Clark, J.I.; Sznol, M.; Logan, T.F.; et al. Ipilimumab in patients with melanoma and brain metastases: An open-label, phase 2 trial. Lancet Oncol. 2012, 13, 459–465.
- Lin, N.U.; Diéras, V.; Paul, D.; Lossignol, D.; Christodoulou, C.; Stemmler, H.J.; Roché, H.; Liu, M.C.; Greil, R.; Ciruelos, E.; et al. Multicenter phase II study of lapatinib in patients with brain metastases from HER2-positive breast cancer. Clin. Cancer Res. 2009, 15, 1452–1459.
- Bachelot, T.; Romieu, G.; Campone, M.; Diéras, V.; Cropet, C.; Dalenc, F.; Jimenez, M.; Le Rhun, E.; Pierga, J.Y.; Gonçalves, A.; et al. Lapatinib plus capecitabine in patients with previously untreated brain metastases from HER2-positive metastatic breast cancer (LANDSCAPE): A single-group phase 2 study. Lancet Oncol. 2013, 14, 64–71.
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, R.M.; Garcia, L.; Yu, L.; et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): Results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014, 15, 1119–1128.
- Costa, D.B.; Shaw, A.T.; Ou, S.H.; Solomon, B.J.; Riely, G.J.; Ahn, M.J.; Zhou, C.; Shreeve, S.M.; Selaru, P.; Polli, A.; et al. Clinical Experience With Crizotinib in Patients with Advanced ALK-Rearranged Non-Small-Cell Lung Cancer and Brain Metastases. J. Clin. Oncol. 2015, 33, 1881–1888.
- Crinò, L.; Ahn, M.J.; De Marinis, F.; Groen, H.J.; Wakelee, H.; Hida, T.; Mok, T.; Spigel, D.; Felip, E.; Nishio, M.; et al. Multicenter Phase II Study of Whole-Body and Intracranial Activity with Ceritinib in Patients With ALK-Rearranged Non-Small-Cell Lung Cancer Previously Treated With Chemotherapy and Crizotinib: Results From ASCEND-2. J. Clin. Oncol. 2016, 34, 2866–2873.
- Iuchi, T.; Shingyoji, M.; Sakaida, T.; Hatano, K.; Nagano, O.; Itakura, M.; Kageyama, H.; Yokoi, S.; Hasegawa, Y.; Kawasaki, K.; et al. Phase II trial of gefitinib alone without radiation therapy for Japanese patients with brain metastases from EGFR-mutant lung adenocarcinoma. Lung Cancer 2013, 82, 282–287.
- Porta, R.; Sánchez-Torres, J.M.; Paz-Ares, L.; Massutí, B.; Reguart, N.; Mayo, C.; Lianes, P.; Queralt, C.; Guillem, V.; Salinas, P.; et al. Brain metastases from lung cancer responding to erlotinib: The importance of EGFR mutation. Eur. Respir. J. 2011, 37, 624–631.
- Schuler, M.; Wu, Y.L.; Hirsh, V.; O’Byrne, K.; Yamamoto, N.; Mok, T.; Popat, S.; Sequist, L.V.; Massey, D.; Zazulina, V.; et al. First-Line Afatinib versus Chemotherapy in Patients with Non-Small Cell Lung Cancer and Common Epidermal Growth Factor Receptor Gene Mutations and Brain Metastases. J. Thorac. Oncol. 2016, 11, 380–390.
- Remon, J.; Steuer, C.E.; Ramalingam, S.S.; Felip, E. Osimertinib and other third-generation EGFR TKI in EGFR-mutant NSCLC patients. Ann. Oncol. 2018, 29, i20–i27.
- Swain, S.M.; Baselga, J.; Miles, D.; Im, Y.H.; Quah, C.; Lee, L.F.; Cortés, J. Incidence of central nervous system metastases in patients with HER2-positive metastatic breast cancer treated with pertuzumab, trastuzumab, and docetaxel: Results from the randomized phase III study CLEOPATRA. Ann. Oncol. 2014, 25, 1116–1121.
- Bartsch, R.; Berghoff, A.S.; Preusser, M. Breast cancer brain metastases responding to primary systemic therapy with T-DM1. J. Neurooncol. 2014, 116, 205–206.
- Taillibert, S.; Chamberlain, M.C. Leptomeningeal metastasis. Handb. Clin. Neurol. 2018, 149, 169–204.
- Jiang, B.-Y.; LI, Y.; Chuai, S.; Zhang, Z.; Yang, J.-J.; Zhong, W.; Zhou, Q.; Wu, Y.-L. NGS to reveal heterogeneity between cerebrospinal fluid and plasma ctDNA among non-small cell lung cancer patients with leptomeningeal carcinomatosis. J. Clin. Oncol. 2017, 35, 9022.
- Zhao, Y.; He, J.Y.; Cui, J.Z.; Meng, Z.Q.; Zou, Y.L.; Guo, X.S.; Chen, X.; Wang, X.; Yan, L.T.; Han, W.X.; et al. Detection of genes mutations in cerebrospinal fluid circulating tumor DNA from neoplastic meningitis patients using next generation sequencing. BMC Cancer 2020, 20, 690.
- Ma, C.; Zhang, J.; Tang, D.; Ye, X.; Li, J.; Mu, N.; Li, Z.; Liu, R.; Xiang, L.; Huang, C.; et al. Tyrosine Kinase Inhibitors Could Be Effective Against Non-small Cell Lung Cancer Brain Metastases Harboring Uncommon EGFR Mutations. Front. Oncol. 2020, 10, 224.
- Kawahara, A.; Abe, H.; Murata, K.; Ishii, H.; Azuma, K.; Takase, Y.; Hattori, S.; Naito, Y.; Akiba, J. Screening system for epidermal growth factor receptor mutation detection in cytology cell-free DNA of cerebrospinal fluid based on assured sample quality. Cytopathology 2019, 30, 144–149.
- Ma, C.; Yang, X.; Xing, W.; Yu, H.; Si, T.; Guo, Z. Detection of circulating tumor DNA from non-small cell lung cancer brain metastasis in cerebrospinal fluid samples. Thorac. Cancer 2020, 11, 588–593.
- Siravegna, G.; Geuna, E.; Mussolin, B.; Crisafulli, G.; Bartolini, A.; Galizia, D.; Casorzo, L.; Sarotto, I.; Scaltriti, M.; Sapino, A.; et al. Genotyping tumour DNA in cerebrospinal fluid and plasma of a HER2-positive breast cancer patient with brain metastases. ESMO Open 2017, 2, e000253.
- Yamamoto, W.; Tomita, N.; Watanabe, R.; Hattori, Y.; Nakajima, Y.; Hyo, R.; Hashimoto, C.; Motomura, S.; Ishigatsubo, Y. Central nervous system involvement in diffuse large B-cell lymphoma. Eur. J. Haematol. 2010, 85, 6–10.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017.
- El-Galaly, T.C.; Cheah, C.Y.; Bendtsen, M.D.; Nowakowski, G.S.; Kansara, R.; Savage, K.J.; Connors, J.M.; Sehn, L.H.; Goldschmidt, N.; Shaulov, A.; et al. Treatment strategies, outcomes and prognostic factors in 291 patients with secondary CNS involvement by diffuse large B-cell lymphoma. Eur. J. Cancer 2018, 93, 57–68.
- Fontanilles, M.; Marguet, F.; Bohers, É.; Viailly, P.J.; Dubois, S.; Bertrand, P.; Camus, V.; Mareschal, S.; Ruminy, P.; Maingonnat, C.; et al. Non-invasive detection of somatic mutations using next-generation sequencing in primary central nervous system lymphoma. Oncotarget 2017, 8, 48157–48168.
- Hattori, K.; Sakata-Yanagimoto, M.; Suehara, Y.; Yokoyama, Y.; Kato, T.; Kurita, N.; Nishikii, H.; Obara, N.; Takano, S.; Ishikawa, E.; et al. Clinical significance of disease-specific MYD88 mutations in circulating DNA in primary central nervous system lymphoma. Cancer Sci. 2018, 109, 225–230.
- Grommes, C.; Tang, S.S.; Wolfe, J.; Kaley, T.J.; Daras, M.; Pentsova, E.I.; Piotrowski, A.F.; Stone, J.; Lin, A.; Nolan, C.P.; et al. Phase 1b trial of an ibrutinib-based combination therapy in recurrent/refractory CNS lymphoma. Blood 2019, 133, 436–445.
- Hiemcke-Jiwa, L.S.; Minnema, M.C.; Radersma-van Loon, J.H.; Jiwa, N.M.; de Boer, M.; Leguit, R.J.; de Weger, R.A.; Huibers, M.M.H. The use of droplet digital PCR in liquid biopsies: A highly sensitive technique for MYD88 p.(L265P) detection in cerebrospinal fluid. Hematol. Oncol. 2018, 36, 429–435.
- Hiemcke-Jiwa, L.S.; Leguit, R.J.; Snijders, T.J.; Bromberg, J.E.C.; Nierkens, S.; Jiwa, N.M.; Minnema, M.C.; Huibers, M.M.H. MYD88 p.(L265P) detection on cell-free DNA in liquid biopsies of patients with primary central nervous system lymphoma. Br. J. Haematol. 2019, 185, 974–977.
- Hickmann, A.K.; Frick, M.; Hadaschik, D.; Battke, F.; Bittl, M.; Ganslandt, O.; Biskup, S.; Döcker, D. Molecular tumor analysis and liquid biopsy: A feasibility investigation analyzing circulating tumor DNA in patients with central nervous system lymphomas. BMC Cancer 2019, 19, 192.
- Rimelen, V.; Ahle, G.; Pencreach, E.; Zinniger, N.; Debliquis, A.; Zalmaï, L.; Harzallah, I.; Hurstel, R.; Alamome, I.; Lamy, F.; et al. Tumor cell-free DNA detection in CSF for primary CNS lymphoma diagnosis. Acta Neuropathol. Commun. 2019, 7, 43.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA Cancer J. Clin. 2016, 66, 7–30.
- Udaka, Y.T.; Packer, R.J. Pediatric Brain Tumors. Neurol. Clin. 2018, 36, 533–556.
- Smoll, N.R.; Drummond, K.J. The incidence of medulloblastomas and primitive neurectodermal tumours in adults and children. J. Clin. Neurosci. 2012, 19, 1541–1544.
- Taylor, M.D.; Northcott, P.A.; Korshunov, A.; Remke, M.; Cho, Y.J.; Clifford, S.C.; Eberhart, C.G.; Parsons, D.W.; Rutkowski, S.; Gajjar, A.; et al. Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 2012, 123, 465–472.
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A.; et al. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317.
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.e736.
- Schwalbe, E.C.; Lindsey, J.C.; Nakjang, S.; Crosier, S.; Smith, A.J.; Hicks, D.; Rafiee, G.; Hill, R.M.; Iliasova, A.; Stone, T.; et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: A cohort study. Lancet Oncol. 2017, 18, 958–971.
- Hovestadt, V.; Ayrault, O.; Swartling, F.J.; Robinson, G.W.; Pfister, S.M.; Northcott, P.A. Medulloblastomics revisited: Biological and clinical insights from thousands of patients. Nat. Rev. Cancer 2020, 20, 42–56.
- Koschmann, C.; Bloom, K.; Upadhyaya, S.; Geyer, J.R.; Leary, S.E. Survival After Relapse of Medulloblastoma. J. Pediatr. Hematol. Oncol. 2016, 38, 269–273.
- Zapotocky, M.; Mata-Mbemba, D.; Sumerauer, D.; Liby, P.; Lassaletta, A.; Zamecnik, J.; Krskova, L.; Kyncl, M.; Stary, J.; Laughlin, S.; et al. Differential patterns of metastatic dissemination across medulloblastoma subgroups. J. Neurosurg. Pediatr. 2018, 21, 145–152.
- Ramaswamy, V.; Remke, M.; Bouffet, E.; Faria, C.C.; Perreault, S.; Cho, Y.J.; Shih, D.J.; Luu, B.; Dubuc, A.M.; Northcott, P.A.; et al. Recurrence patterns across medulloblastoma subgroups: An integrated clinical and molecular analysis. Lancet Oncol. 2013, 14, 1200–1207.
- Hill, R.M.; Kuijper, S.; Lindsey, J.C.; Petrie, K.; Schwalbe, E.C.; Barker, K.; Boult, J.K.; Williamson, D.; Ahmad, Z.; Hallsworth, A.; et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer Cell 2015, 27, 72–84.
- Sun, Y.; Li, M.; Ren, S.; Liu, Y.; Zhang, J.; Li, S.; Gao, W.; Gong, X.; Liu, J.; Wang, Y.; et al. Exploring genetic alterations in circulating tumor DNA from cerebrospinal fluid of pediatric medulloblastoma. Sci. Rep. 2021, 11, 5638.
- Hovestadt, V.; Jones, D.T.; Picelli, S.; Wang, W.; Kool, M.; Northcott, P.A.; Sultan, M.; Stachurski, K.; Ryzhova, M.; Warnatz, H.J.; et al. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature 2014, 510, 537–541.
- Li, J.; Zhao, S.; Lee, M.; Yin, Y.; Li, J.; Zhou, Y.; Ballester, L.Y.; Esquenazi, Y.; Dashwood, R.H.; Davies, P.J.A.; et al. Reliable tumor detection by whole-genome methylation sequencing of cell-free DNA in cerebrospinal fluid of pediatric medulloblastoma. Sci. Adv. 2020, 6.