Aldose reductase (AR, ALR2), the first enzyme of the polyol pathway, is implicated in the pathophysiology of diabetic complications. Aldose reductase inhibitors (ARIs) thus present a promising therapeutic approach to treat a wide array of diabetic complications.
- indole
- pyridoindole
- triazinoindole
- aldose reductase
- inhibitor
- diabetic complications
- polyol pathway
- antioxidant
1. Introduction
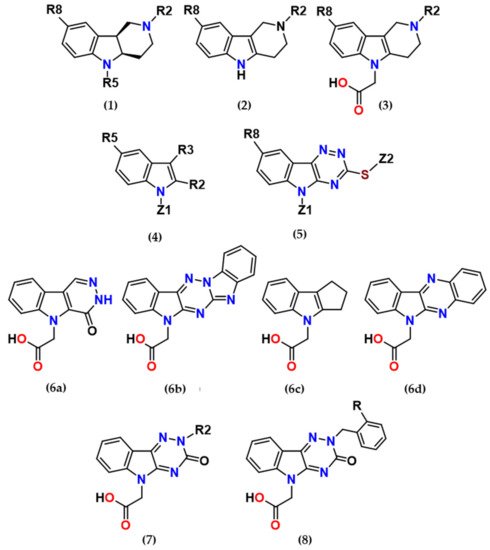
2. Main Results and Future Perspectives
several promising series of aldose reductase inhibitors/antioxidants as summarized in Figure 2.
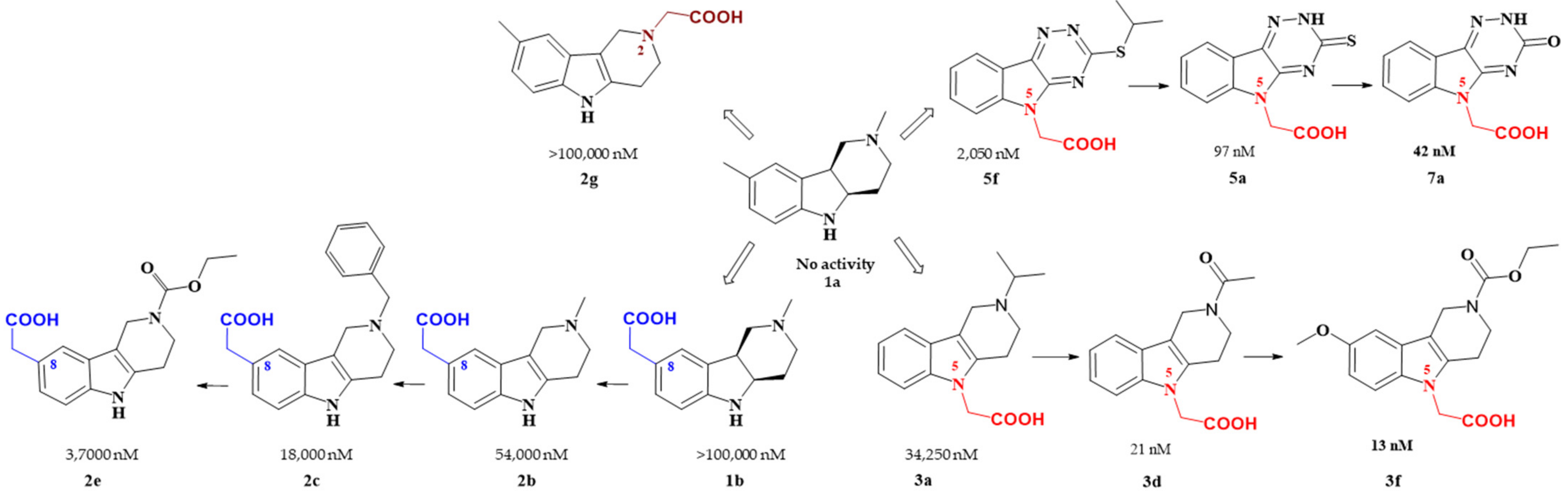
Figure 2. Carboxymethyl pharmacophore regioisomerization in AR inhibitory activity optimization starting from the non-active hexahydropyridoindole scaffold 1a
Starting from the efficient hexahydropyridoindole antioxidant stobadine (1a), two series of hexahydro- and tetrahydropyridoindoles carboxymethylated in position 8 were synthesized and characterized as AR inhibitors with negligible (e.g. compound 1b) or mild (e.g. compounds 2b,c) efficacy and selectivity yet with significant antioxidant (AO) effect as an additional biological activity. The hexahydropyridoindole scaffold was excluded from further drug design since the marked antioxidant activity of the hexahydropyridoindoles was combined with only minor AR inhibition (e.g. compound 1b) which was explained by space distortion of the hexahydropyridoindole structure not fitting properly the AR binding pocket. Tetrahydropyridoindole congeners with a planar tricyclic moiety appeared more promising route in designing efficient ARIs. Notably, elimination of the basicity center at N2 position, which prevented zwitterions formation, significantly improved AR inhibition as shown in the case of compound 2e.
Shifting the carboxymethyl pharmacophore from position 8 to position 2, yielded derivatives with markedly decreased inhibition efficacy, as exemplified by compound 2g, therefore this route of drug designing was not further followed.
Structure optimization of the tetrahydropyridoindole scaffold by transferring the carboxymethyl pharmacophore from position 8 to position 5, yielded derivatives with markedly enhanced inhibition efficacy and selectivity, yet with abolished AO activity (e.g. compounds 3a,d,f). In this series, the AR inhibition efficacy markedly increased with decreasing basicity of N2 nitrogen as documented by compound 3f.
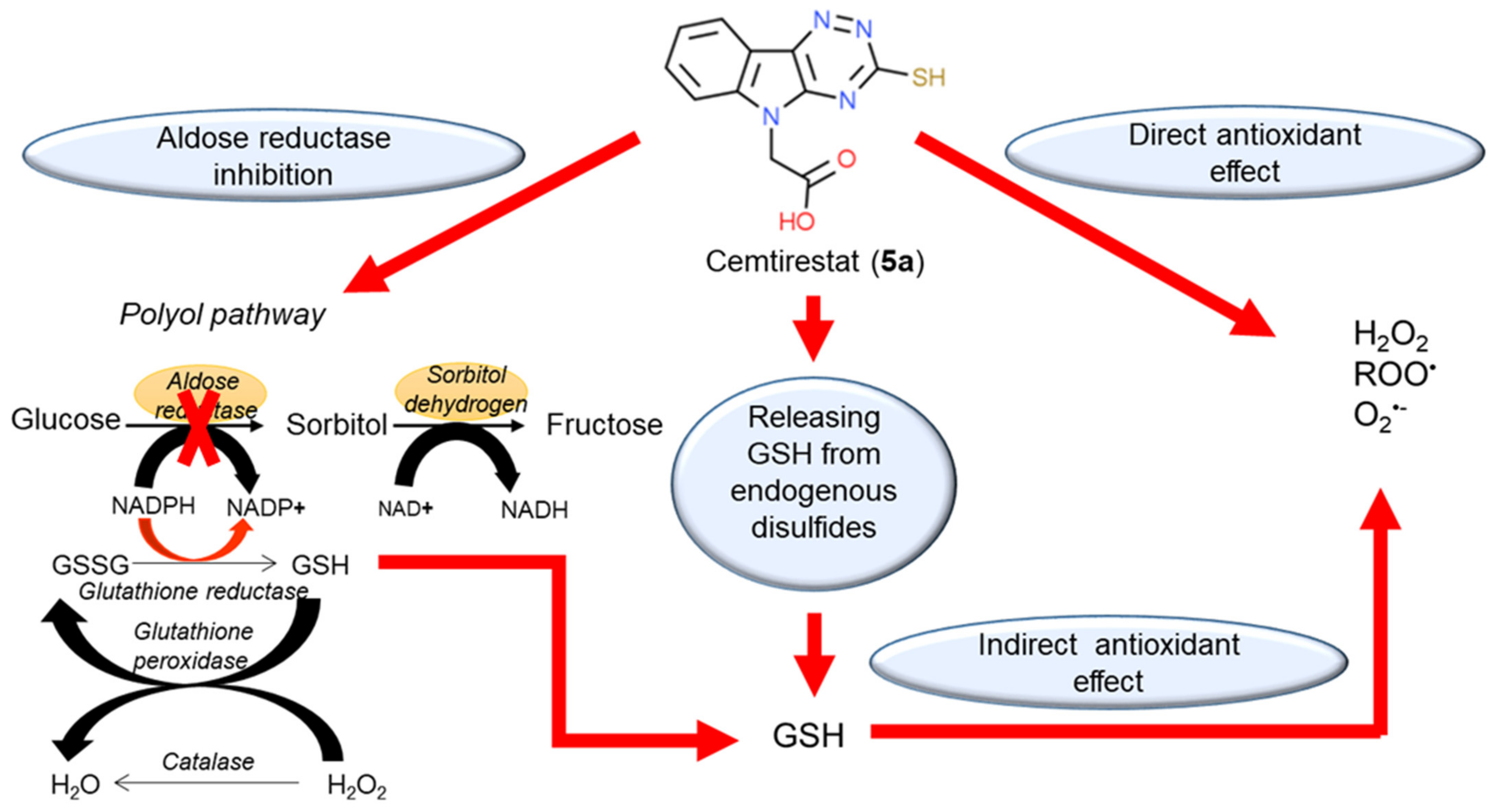
Thioxotriazine structural alternatives yielded highly efficient AR inhibitors (e.g. compound 5a) with high selectivity and reasonable AO activity. In further structure optimization efforts, isosteric replacement of sulfur in compound 5a with oxygen provided compound 7a with increased AR inhibition efficacy and markedly improved selectivity, yet with diminished AO activity.
Interestingly, the structural features of the most efficient ARIs resulting from the above optimization procedure, namely compounds 2c, 3f, 5a and 7a (IC50(ALR2) = 18 000; 13; 97; 42 nM, resp.) still match the strict criteria of the „rule of three“, which points to their excellent “leadlikeness” with prospects of further structure optimizations.
3. Patents
References
- Kador, P.; Lee, J.; Fujisawa, S.; Blessing, K.; Lou, M. Relative Importance of Aldose Reductase versus Nonenzymatic Glycosylation on Sugar Cataract Formation in Diabetic Rats. J. Ocul. Pharmacol. Ther. 2000, 16, 149–160.
- Oates, P. Aldose Reductase, Still a Compelling Target for Diabetic Neuropathy. Curr. Drug Targets. 2008, 9, 14–36.
- Alexiou, P.; Pegklidou, K.; Chatzopoulou, M.; Nicolaou, I.; Demopoulos, V. Aldose Reductase Enzyme and Its Implication to Major Health Problems of the 21(st) Century. Curr. Med. Chem. 2009, 16, 734–752.
- Maccari, R.; Ottanà, R. Targeting Aldose Reductase for the Treatment of Diabetes Complications and Inflammatory Diseases: New Insights and Future Directions. J. Med. Chem. 2015, 58, 2047–2067.
- Hotta, N. New Approaches for Treatment in Diabetes: Aldose Reductase Inhibitors. Biomed. Pharmacother. 1995, 49, 232–243.
- Costantino, L.; Rastelli, G.; Vianello, P.; Cignarella, G.; Barlocco, D. Diabetes Complications and Their Potential Prevention: Aldose Reductase Inhibition and Other Approaches. Med. Res. Rev. 1999, 19, 3–23.
- Grewal, A.; Bhardwaj, S.; Pandita, D.; Lather, V.; Sekhon, B. Updates on Aldose Reductase Inhibitors for Management of Diabetic Complications and Non-Diabetic Diseases. Mini Rev. Med. Chem. 2016, 16, 120–162.
- Miyamoto, S. Recent advances in aldose reductase inhibitors: Potential agents for the treatment of diabetic complications. Expert Opin. Ther. Patents 2002, 12, 621–631.
- Chatzopoulou, M.; Alexiou, P.; Kotsampasakou, E.; Demopoulos, V. Novel Aldose Reductase Inhibitors: A Patent Survey (2006--Present). Expert Opin. Ther. Pat. 2012, 22, 1303–1323.
- Quattrini, L.; La Motta, C. Aldose Reductase Inhibitors: 2013-Present. Expert Opin. Ther. Pat. 2019, 29, 199–213.
- Kousaxidis, A.; Petrou, A.; Lavrentaki, V.; Fesatidou, M.; Nicolaou, I.; Geronikaki, A. Aldose reductase and protein tyrosine phosphatase 1B inhibitors as a promising therapeutic approach for diabetes mellitus. Eur. J. Med. Chem. 2020, 207, 112742.
- Kumar, M.; Choudhary, S.; Singh, P.; Silakari, O. Addressing Selectivity Issues of Aldose Reductase 2 Inhibitors for the Management of Diabetic Complications. Future Med. Chem. 2012, 12, 1327–1358.
- Khayami, R.; Hashemi, S.; Kerachian, M. Role of Aldo-Keto Reductase Family 1 Member B1 (AKR1B1) in the Cancer Process and Its Therapeutic Potential. J. Cell. Mol. Med. 2020, 24, 8890–8902.
- Liu, J.; Wen, G.; Cao, D. Aldo-Keto Reductase Family 1 Member B1 Inhibitors: Old Drugs with New Perspectives. Recent Pat. Anticancer Drug Discov. 2009, 4, 246–253.
- Taskoparan, B.; Seza, E.; Demirkol, S.; Tuncer, S.; Stefek, M.; Gure, A.; Banerjee, S. Opposing Roles of the Aldo-Keto Reductases AKR1B1 and AKR1B10 in Colorectal Cancer. Cell. Oncol. 2017, 40, 563–578.
- Chatzopoulou, M.; Pegklidou, K.; Papastavrou, N.; Demopoulos, V.J. Development of aldose reductase inhibitors for the treatment of inflammatory disorders. Expert Opin. Drug Discov. 2013, 8, 1365–1380.
- Del Corso, A.; Cappiello, M.; Mura, U. From a Dull Enzyme to Something Else: Facts and Perspectives Regarding Aldose Reductase. Curr. Med. Chem. 2008, 15, 1452–1461.
- Ramana, K.; Srivastava, S. Aldose Reductase: A Novel Therapeutic Target for Inflammatory Pathologies. Int. J. Biochem. Cell. Biol. 2010, 24, 17–20.
- Morava, E. Elevated Sorbitol Underlies a Heritable Neuropathy. Nat. Genet. 2020, 52, 469–470.
- Baynes, J.; Thorpe, S. Role of Oxidative Stress in Diabetic Complications: A New Perspective on an Old Paradigm. Diabetes. Diabetes 1999, 48, 1–9.
- Brownlee, M. Biochemistry and Molecular Cell Biology of Diabetic Complications. Nature 2001, 414, 813–820.
- Nishikawa, T.; Edelstein, D.; Brownlee, M. The Missing Link: A Single Unifying Mechanism for Diabetic Complications. Kidney Int. Suppl. 2000, 77, 26–30.
- Obrosova, I. How Does Glucose Generate Oxidative Stress in Peripheral Nerve? Int. Rev. Neurobiol. 2002, 50, 3–35.
- Tomlinson, D.; Gardiner, N. Glucose Neurotoxicity. Nat. Rev. Neurosci. 2008, 9, 36–45.
- Aldini, G.; Vistoli, G.; Stefek, M.; Chondrogianni, N.; Grune, T.; Sereikaite, J.; Sadowska-Bartosz, I.; Bartosz, G. Molecular Strategies to Prevent, Inhibit, and Degrade Advanced Glycoxidation and Advanced Lipoxidation End Products. Free Radic. Res. 2013, 47, 93–137.
- De Sá Alves, F.; Barreiro, E.; Fraga, C. From nature to drug discovery: The indole scaffold as a ‘privileged structure’. Mini Rev. Med. Chem. 2009, 9, 782–793.
- Welsch, M.; Snyder, S.; Stockwell, B. Privileged Scaffolds for Library Design and Drug Discovery. Curr. Opin. Chem. Biol. 2010, 14, 341–361.
- Thanikachalam, P.; Maurya, R.; Garg, V.; Monga, V. An Insight into the Medicinal Perspective of Synthetic Analogs of Indole: A Review. Eur. J. Med. Chem. 2019, 15, 562–612.
- Kumari, A.; Singh, R.K. Medicinal Chemistry of Indole Derivatives: Current to Future Therapeutic Prospectives. Bioorg. Chem. 2019, 103021.
- Taliani, S.; Da Da Settimo, F.; Martini, C.; Laneri, S.; Novellino, E.; Greco, G. Exploiting the Indole Scaffold to Design Compounds Binding to Different Pharmacological Targets. Molecules 2020, 25, 2331.
- Juranek, I.; Horakova, L.; Rackova, L.; Stefek, M. Antioxidants in Treating Pathologies Involving Oxidative Damage: An Update on Medicinal Chemistry and Biological Activity of Stobadine and Related Pyridoindoles. Curr. Med. Chem. 2010, 17, 552–570.
- Juránek, I.; Račková, L.; Stefek, M. Stobadine – An Indole Type Alternative to the Phenolic Antioxidant Reference Trolox. Biochemistry 2012, 443–452.
- Kovacikova, L.; Majekova, M.; Stefek, M. Substituted Pyridoindoles as Biological Antioxidants: Drug Design, Chemical Synthesis, and Biological Activity. Methods Mol. Biol. 2015, 1208, 313–327.
- Augustyniak, A.; Bartosz, G.; Cipak, A.; Duburs, G.; Horáková, L.; Luczaj, W.; Majekova, M.; Odysseos, A.; Rackova, L.; Skrzydlewska, E.; et al. Natural and Synthetic Antioxidants: An Updated Overview. Free Radic. Res. 2010, 44, 1216–1262.
- Stefek, M.; Gajdosik, A.; Gajdosikova, A.; Juskova, M.; Kyselova, Z.; Majekova, M.; Rackova, L.; Snirc, V.; Demopoulos, V.; Karasu, C. Stobadine and Related Pyridoindoles as Antioxidants and Aldose Reductase Inhibitors in Prevention of Diabetic Complications. In Advances in Molecular Mechanisms and Pharmacology of Diabetic Complications; Stefek, M., Ed.; Transworld Research Network: Kerala, India, 2010; pp. 285–307.
- Van Zandt, M.; Jones, M.; Gunn, D.; Geraci, L.; Jones, J.; Sawicki, D.; Sredy, J.; Jacot, J.; Dicioccio, A.; Petrova, A.; et al. Discovery of 3-[(4,5,7-Trifluorobenzothiazol-2-Yl)Methyl]Indole-N-Acetic Acid (Lidorestat) and Congeners as Highly Potent and Selective Inhibitors of Aldose Reductase for Treatment of Chronic Diabetic Complications. J. Med. Chem. 2005, 48, 3141–3152.
- Stefek, M.; Soltesova Prnova, M.; Majekova, M.; Rechlin, C.; Heine, A.; Klebe, G. Identification of Novel Aldose Reductase Inhibitors Based on Carboxymethylated Mercaptotriazinoindole Scaffold. J. Med. Chem. 2015, 58, 2649–2657.
- Zhan, J.; Ma, K.; Zheng, Q.; Yang, G.; Zhang, H. Exploring the Interactional Details between Aldose Reductase (AKR1B1) and 3-Mercapto-5H-1,2,4-Triazino[5,6-b]Indole-5-Acetic Acid through Molecular Dynamics Simulations. J. Biomol. Struct. Dyn. 2019, 37, 1724–1735.
- Prnova, M.S.; Ballekova, J.; Majekova, M.; Stefek, M. Antioxidant Action of 3-Mercapto-5h-1,2,4-Triazino[5,6-b]Indole-5-Acetic Acid, an Efficient Aldose Reductase Inhibitor, in a 1,1′-Diphenyl-2-Picrylhydrazyl Assay and in the Cellular System of Isolated Erythrocytes Exposed to Tert-Butyl Hydroperoxide. Redox Rep. 2015, 20, 282–288.
- Štefek, M.; Šoltésová Prnová, M.; Balleková, J.; Májeková, M. Cemtirestat, a Novel Aldose Reductase Inhibitor and Antioxidant, in Multitarget Pharmacology of Diabetic Complications. In Proceedings of the Research World International Conference, Muscat, Oman, 20 May 2016; pp. 1–4, ISBN 978-93-86083-34-0.
- Soltesova Prnova, M.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Colín-González, A.L.; Piedra-García, F.; Rangel-López, E.; Kovacikova, L.; Ceylan, A.; Karasu, C.; Santamaria, A.; et al. Antioxidant Mechanisms in the Neuroprotective Action of Cemtirestat: Studies in Chemical Models, Liposomes and Rat Brain Cortical Slices. Neuroscience 2020, 443, 206–217.
- Soltesova Prnova, M.; Ballekova, J.; Gajdosikova, A.; Gajdosik, A.; Stefek, M. A Novel Carboxymethylated Mercaptotriazinoindole Inhibitor of Aldose Reductase Interferes with the Polyol Pathway in Streptozotocin-Induced Diabetic Rats. Physiol. Res. 2015, 64, 587–591.
- Prnova, M.S.; Kovacikova, L.; Svik, K.; Bezek, S.; Elmazoğlu, Z.; Karasu, C.; Stefek, M. Triglyceride-Lowering Effect of the Aldose Reductase Inhibitor Cemtirestat—Another Factor That May Contribute to Attenuation of Symptoms of Peripheral Neuropathy in STZ-Diabetic Rats. Naunyn. Schmiedebergs Arch. Pharmacol. 2020, 393, 651–661.
- Valachová, K.; Mach, M.; Šoltés, L. Oxidative Degradation of High-Molar-Mass Hyaluronan: Effects of Some Indole Derivatives to Hyaluronan Decay. Int. J. Mol. Sci. 2020, 21, 5609.
- Elmazoglu, Z.; Prnova, M.S.; Santamaria, A.; Stefek, M.; Karasu, C. Combatting Nitrosative Stress and Inflammation with Novel Substituted Triazinoindole Inhibitors of Aldose Reductase in PC12 Cells Exposed to 6-Hydroxydopamine Plus High Glucose. Neurotox. Res. 2021, 39, 210–226.
- Elmazoglu, Z.; Prnova, M.S.; Stefek, M.; Ceylan, A.F.; Aschner, M.; Rangel-López, E.; Santamaria, A.; Karasu, C. Protective Effects of Novel Substituted Triazinoindole Inhibitors of Aldose Reductase and Epalrestat in Neuron-like PC12 Cells and BV2 Rodent Microglial Cells Exposed to Toxic Models of Oxidative Stress: Comparison with the Pyridoindole Antioxidant Stobadine. Neurotox. Res. 2021.
- Prnová, M.Š.; Račková, L.; Kováčiková, L.; Balleková, J.; Viskupičová, J.; Micháliková, S.; Taşkoparan, B.; Elmazoğlu, Z.; Rižner, T.L.; Karasu, C.; et al. General toxicity assessment of the novel aldose reductase inhibitor cemtirestat. Interdiscip. Toxicol. 2019, 12, 120–128.
- Soltesova Prnova, M.; Svik, K.; Bezek, S.; Kovacikova, L.; Karasu, C.; Stefek, M. 3-Mercapto-5H-1,2,4-Triazino[5,6-b]Indole-5-Acetic Acid (Cemtirestat) Alleviates Symptoms of Peripheral Diabetic Neuropathy in Zucker Diabetic Fatty (ZDF) Rats: A Role of Aldose Reductase. Neurochem. Res. 2019, 44, 1056–1064.