1. Introduction
Bacteria as microbial pathogens are causative agents of life-threatening infectious diseases [1]. Such pathogenic bacteria produce alarming numbers in terms of morbidity and mortality outcomes [2][3]. One crucial avenue towards bacterial pathogenesis involves the reduction in the therapeutic effects of antibacterial chemotherapy [4][5]. Throughout their evolutionary history, bacterial pathogens have developed various means of resisting the inhibitory and bactericidal consequences of antimicrobial agents [4]. Such antimicrobial resistance systems involve the engagement of bacterial molecular and cellular-based machinery [6]. Interestingly, the selection of a bacterial variant with resistance to a single antimicrobial agent frequently manifests the emergence of a multidrug resistance characteristic in the new mutant [7]. Newly emerged bacterial pathogens with resistance to multiple antibacterial agents can result in compromised efficacy in the treatment of infection [3][8].
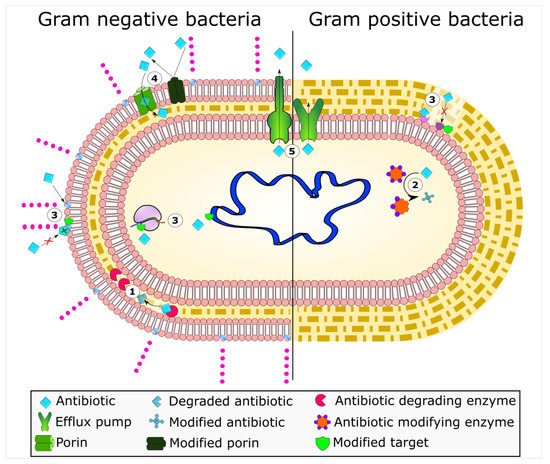
Mechanisms of antimicrobial resistance include the active export systems within the membranes of bacteria, prevention of antimicrobial entrance into cells of pathogenic bacteria, enzymatic destruction of antimicrobial agents, production of thick biofilms, modified targets of antimicrobials, and bacterial sites of action that are protected from antimicrobials, () [2][4]. Furthermore, multidrug-resistant bacteria have developed mechanisms that confer the DNA transfer of genetic determinants of resistance to pathogenic species in the clinical setting, the food production industry, the human gut, and in agriculture [9].
Figure 1. Bacterial mechanisms of resistance to antimicrobial agents. The common mechanisms of antibiotic resistance in bacteria are enzymatic hydrolysis (1), enzymatic modifications of antibiotics by group transfer and redox process (2), modifications of antibiotic targets (3), reduced permeability to antibiotics by modifications of porins (4), and active extrusion of antibiotics by membrane efflux pumps (5).
Thus, new strategies for the circumvention of bacterial resistance to antimicrobial agents are desired [10]. In order to discover novel approaches to address multiple antimicrobial resistances in these microbial pathogens, however, it is necessary to attain a clear understanding of these resistance systems at the molecular and cellular levels. For young and new investigators, here, we consider an introductory overview of each of these disparate bacterial resistance mechanisms here.
2. Enzyme-Based Antimicrobial-Inactivation Systems
Along the timeline of antibiotic discovery and introduction, several enzymatic mechanisms of antibiotic inactivation were also discovered. Although very few novel mechanisms of antibiotic resistance have been reported in recent times, several new variants of known enzymes that endow bacteria with resistance to newly introduced drugs have emerged, suggesting that the bacterial response to new antibiotics or the modified versions of existing antibiotics is swift. The enzymatic mechanisms of antibiotic resistance include hydrolysis, group transfer, and redox processes [4]. In terms of diversity, evolution, and spread, antibiotic resistance enzymes contribute remarkably to the bacterial ability to overcome antibiotic pressure. The β-lactamases are the oldest known and the most diverse antibiotic degrading enzymes that cleave the β-lactam ring of the penicillin group of antibiotics and render them ineffective. The first such β-lactamase was discovered soon after the first antibiotic penicillin was in clinical use. Scientific evidence suggests the existence of β-lactamases before penicillin was clinically employed, emphasizing that the production of antimicrobial compounds and the mechanisms to endure them occur in parallel in the environment [11]. Bacteria that produce antibiotics apparently require mechanisms to overcome the lethal effects of the compounds, and these are in the form of concurrent production of degradative enzymes, mutations in targets of antibiotics, or active extrusion of antibiotics from the cell so that the antibiotic-producing cell is protected. However, the selection pressure created due to the extensive use of antibiotics in humans and animals propagated the resistant clones of bacteria in clinical and food production environments. In due course of time, genetic exchange mechanisms facilitated the wider dissemination of resistance traits in bacterial communities. The introduction of more antibiotics, newer as well modified, augmented the process of evolution and spread of resistance mechanisms. Since the majority of the antibiotics introduced in the last two decades are mostly the modified versions of existing antibiotics belonging to the same classes (e.g., β-lactams), a few mutations in the enzymes could render bacteria quickly resistant to them [11].
The β-lactams constitute the largest group of clinically used antibiotics, comprising of penicillins, cephalosporins of different generations, monobactams, and carbapenems, all of which are characterized by the presence of 3-carbon, 1-nitrogen containing β-lactam ring. The β-lactam antibiotics inhibit the bacterial proteins known as penicillin-binding proteins (PBPs), which perform the critical role of peptide cross-linking during peptidoglycan cell wall biosynthesis. The structural mimicry of the d-Ala-d-Ala terminal fragment of cross-linking peptide by β-lactams facilitates competitive inhibition of PBPs [12], which stops the cell wall synthesis leading to bacterial cell lysis and death [13]. However, bacteria gain resistance to lactam antibiotics by modifying their PBPs, which are no longer susceptible to binding by the antibiotic. Alternatively, bacteria produce powerful lactamases that degrade antibiotics before they can bind with the PBPs. Since their discovery in the early 1940s, the family of β-lactamases has grown seamlessly, with more than 300 enzymes identified globally [14][15].
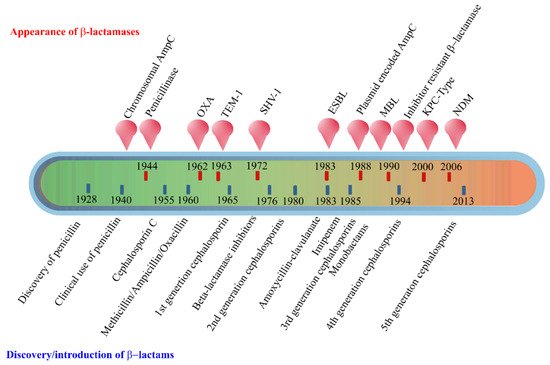
The early β-lactamases were penicillinase enzymes that degraded penicillin, which started appearing rapidly in clinical bacteria [16][17]. The introduction of modified, semisynthetic penicillins such as methicillin, ampicillin, and amoxicillin resulted in the gradual appearance of β-lactamases capable of degrading them. The first plasmid-borne transferrable β-lactamase was TEM-1, followed by TEM-2 and SHV-1 enzymes [18][19]. TEM is the most common mechanism of ampicillin resistance compared to less prevalent SHV-1, although both have the same affinity for this antibiotic. TEM and SHV share 60% amino acid similarity between them and are inhibited by clavulanic acid, tazobactam, and sulbactam. The discovery of cephalosporin C in the early 1960s heralded an era of synthetic cephalosporins, which was thought to fend off β-lactamases. Structurally, cephalosporins have their β-lactam ring fused to a six-membered dihydrothiazine ring compared to penicillins in which the β-lactam is fused with a five-membered thiazolidine ring [20]. Subsequently, carbapenem and monobactam groups of β-lactam antibiotics with structurally variant lactam rings were discovered from natural sources and formed the basis for the synthesis of similar compounds with modifications. However, the enzymes extended-spectrum β-lactamases (ESBLs) that could hydrolyze a wide range of cephalosporins emerged from TEM and SHV lactamases by point mutations [18]. ESBLs hydrolyze a broad spectrum of cephalosporins, including first, second, third-generation cephalosporins and aztreonam, but not cephamycins and carbapenems, and are inhibited by clavulanic acid [18][21]. As a consequence of mutations and the expansion of the substrate range, ESBLs have a lesser affinity for classical β-lactams compared to their ancestral β-lactamases. Subsequently, CTX-M type ESBLs with high affinity for cefotaxime emerged independent of TEM and SHV lactamases, and these supposedly evolved from β-lactamases of Kluyvera spp. [22]. Over the years, CTX-M has overtaken other ESBLs in terms of number and global distribution, with more than 230 types identified to date. shows the timeline of the evolution of β-lactamases in relation to the introduction of β-lactam antibiotics for clinical use.
Figure 2. Evolution of β-lactamases. Within five decades of discovering the first penicillin-degrading enzyme, β-lactamases capable of hydrolyzing most β-lactam antibiotics, and resistance to inhibitors have emerged. The ability to tolerate a broad spectrum of β-lactams and inhibitor combinations is bolstered by the presence of multiple β-lactamase-encoding genes in a single pathogen.
The initial efforts to classify β-lactamases were based on their functional characteristics such as the substrate-inhibitor profiles, protein molecular weight, isoelectric point, etc. [12][14][23]. A second approach employed amino acid sequence similarities and enzymatic activities to classify β-lactamases into four main groups, of which groups A, C, and D are serine β-lactamases, while class B is composed of metallo β-lactamases that require active site zinc ion(s) for their hydrolytic activities [12][24]. Group A enzymes form the largest group of lactamases comprising some of the critical resistance enzymes such as TEM, SHV, and CTX-M type of β-lactamases. Other important ESBLs include the carbapenem hydrolyzing KPC type ESBLs originally reported from Klebsiella pneumoniae, which have an expanded substrate spectrum encompassing the cephalosporins and carbapenems but susceptible to inhibition by clavulanates and boronic acid [23][25]. The chromosomally encoded AmpC (class C) cephalosporinases described early in the timeline of the discovery of β-lactamases have no homology with penicillinases and thus constitute a distinct group of enzymes [26][27]. Commonly found in Enterobacteriaceae, AmpC enzymes are inducible and are produced at low basal levels, and preferentially hydrolyze cephalosporins including cefoxitin but not cefepime. These are generally resistant to inhibition by clavulanic acid, sulbactam, or tazobactam. The metallo-β-lactamases or MBLs belonging to class B have vigorous hydrolytic activities against carbapenems and are also active against a range of cephalosporins [28][29]. In 2009, a new variant New Delhi Metallo-β-lactamase (NDM), emerged, and since then, it has been reported from all over the world [29]. NDM confers resistance to all β-lactam antibiotics except aztreonam, and the plasmid carrying blaNDM gene harbors resistance markers for several other antibiotics. VIM and IMP are other important class B carbapenemases commonly encountered in Enterobacteriaceae.
The OXA type enzymes belonging to the Class D lactamase group were originally discovered as plasmid-encoded oxacillin hydrolyzing enzymes in lactose non-fermenting bacteria such as Pseudomonas, Acinetobacter, and Shewanella, and later in Enterobacteriaceae through plasmid exchange [30][31]. These enzymes are poorly inhibited by lactamase inhibitors such as clavulanic acid. Although OXA lactamases have a narrow substrate range composed of penicillins, cloxacillin, and oxacillin, the enzymes evolved to hydrolyze extended-spectrum cephalosporins and carbapenems through point mutations, and these abilities vary among different OXA types [28][32].
The β-lactamase mediated antimicrobial resistance is widespread among ESKAPE (Enterococcus, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and E. coli) group of organisms, infections with which are usually associated with a significantly higher economic burden and highest risk of mortalities [33][34]. The World Health Organization (WHO) has recognized carbapenem-resistant Enterobacteriaceae (CRE) as a serious global health scourge for which the development of new antimicrobials is critically needed [35].
Enzymatic hydrolysis is also a common mechanism of resistance against macrolides, rifampicin, and fosfomycin. Many Enterobacteriaceae members produce plasmid-encoded esterases EreA and EreB that hydrolyze the macrolactone ring of 14- and 15-membered macrolides such as erythromycin A, clarithromycin, and azithromycin [36][37]. The structurally altered macrolide antibiotic will no longer be able to bind to its preferred target site in the ribosome [38].
Another important mechanism of enzymatic degradation is associated with the manganese ion (Mn2+)-dependent, chromosomally-encoded FosX that uses water to cleave the epoxide ring of fosfomycin. Other fosfomycin modifying metalloenzymes include FosA, FosB, and two epoxide kinases FomA and FomB [39]. FosA is a Mn2+ and K+-dependent glutathione-S-transferase, while FosB is a Mg2+ thiol-S-transferase. The mechanism involves adding glutathione or thiol groups to the oxirane ring of fosfomycin resulting in an inactive drug [40]. FomA and FomB kinases utilize ATP and Mn2+ ions to phosphorylate the oxirane ring of fosfomycin [39].
Tetracyclines are in use for over 70 years as widely used antibiotics in human and animal medicine [41]. Tetracycline is broken down by a monooxygenase enzyme Tet(X), which is oxygen- and FAD-dependent [42]. Tet(X) monohydroxylates break down tetracyclines at position 11a, followed by non-enzymatic degradation. Similarly, enzymatic monoxygenation of the naphthyl group of rifamycin antibiotics by monooxygenases (Rox) inactivate them by leading to the linearization of the naphthoquinone or naphthohydroquinone ring [43].
Enzymatic modification of antibiotics by the transfer of functional groups, such as acyl, glycosyl, ribosyl, nucleotidyl, phosphoryl, or thiol groups, confers resistance to a range of antibiotics, including aminoglycosides, rifamycins, macrolides, epoxides, and chloramphenicol [44]. The aminoglycoside modifying enzymes (AME) responsible for resistance to different aminoglycoside antibiotics include N-acetyltransferases (AAC), O-adenyltransferases (ANT), and O-phosphotransferases (APH). These enzymes catalyze the modification of various hydroxyl or the amino groups of the aminoglycosides resulting in their inability to bind to their 30S ribosomal targets [45]. Similarly, in Gram-negative bacteria, a plasmid-encoded ADP-ribosyltransferase (Arr-2) is commonly responsible for rifampin resistance [46]. Similarly, chloramphenicol is modified by acetyl-CoA-dependent acetylation of its 3-hydroxyl group by chloramphenicol acetyltransferase (CAT) enzymes [47]. The modified antibiotic does not bind to its target site, the 50S subunit of ribosomes. CATs are widely distributed among Gram-positive and -negative bacteria and show little amino acid sequence similarities, with only 25 amino acid residues conserved among all CAT variants [47].
3. Alteration of Antimicrobial Targets
As bacterial enzymes mentioned above alter drug structures, the drug targets may likewise be altered, preventing drug binding and, thus, conferring resistance. Antimicrobial targets play vital roles in microbial growth or survival and, thus, serve as potentially useful targets for mitigating infection. In addition, these targets must differ or be completely absent from humans or the animal species being treated with an antimicrobial to allow for a selective mode of action. A classic example of such a target is peptidoglycan. Peptidoglycan is essential to the growth and survival of many bacterial species and has a chemical structure that is not present in the mammalian hosts they infect. This allows for the targeting of enzymes responsible for the synthesis and assembly of peptidoglycan. The function of proteins associated with these target sites makes it non-viable for a bacterium to evolve resistance by removing these proteins. However, mutations that allow for continued functionality while reducing the ability of an antimicrobial agent to bind them at the target site have been a veritable regularity in the arms race between antimicrobial substances and antimicrobial-resistant bacteria. In addition to peptidoglycan, alteration in target sites has been attributed to ribosomes, nucleic acid enzymes, and lipopolysaccharides [48].
As discussed previously in this review, peptidoglycan inhibition by glycopeptides involves the binding of the peptidyl-d-alanyl-d-alanine terminus of peptidoglycan precursors. This binding prevents integration via the transglycosylase activity of these precursors into the cell wall [49], as shown in .
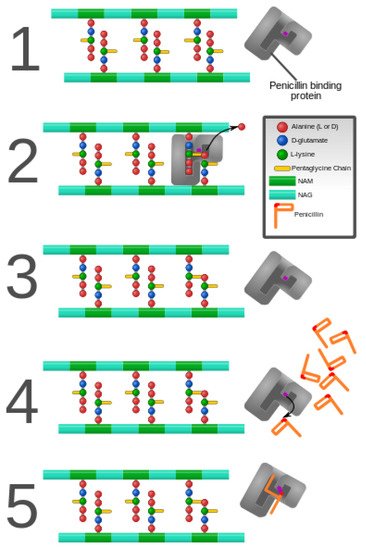
Figure 3. Penicillin and penicillin-binding protein of the bacterial cell wall. (1) The peptidoglycan layer of a bacterial cell wall harbors the repeating moieties of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM). The NAM subunits bind short variable peptide chains, usually l-Ala and two distal d-Ala residues. (2) The PBP cross-links the peptide side chain, releasing a free Ala. (3) Upon cross-linking, the PBP dissociates from the cell wall. (4) Penicillin binds the PBP active site, affecting its enzyme activity. (5) The β-lactam ring of penicillin is cleaved during its reaction with PBP. Penicillin stays covalently bound PBP, permanently inhibiting the active site. Altered PBPs, such as PBP2a, are unable to accommodate penicillin-binding, preventing cell wall synthesis inhibition [48][49].
PCBs are one mechanism for antimicrobial resistance, but the peptidoglycan precursors themselves can undergo alteration, which reduces the affinity of antimicrobials without the involvement of enzymatic inactivation. Such is the case with Enterococcus faecium and E. faecalis, which have been discussed in the literature as developing resistance by acquiring one of two related gene clusters encoding VanA and VanB [50][51]. These gene clusters produce a modified terminus that contains d-alanyl-d-lactate as opposed to d-alanyl-d-alanine [50]. This alteration leads to glycopeptides having a much lower binding affinity [52]. Thus, these gene clusters, found on transposable elements, have allowed the spread of modified targets in enterococci. Similarly, there are rarer but related gene clusters that have been shown to modify peptidoglycan precursors, such as those encoding VanD [53], VanE [54], and Van G [55].
Ribosomes, serving the vital role of protein synthesis, are common to both prokaryotic and eukaryotic organisms but differ quite vastly from one another in structure, making them another suitable candidate for antimicrobial targeting [56]. The 50S ribosomal unit serves as the binding site for macrolide, lincosamide, and streptogramin B [57]. Recalcitrance to these specific antimicrobials is known as MLS(B) type resistance [57], and it results from a post-transcriptional modification of the 23S rRNA component of the 50S ribosomal subunit that is involved with methylation or dimethylation of key adenine bases in the peptidyl transferase functional domain [58]. Mutations in the 23S rRNA, close to the site of methylation have also been associated with resistance to the macrolide group of antibiotics in a range of organisms, such as Helicobacter pylori [59] and propionibacteria [60]. Macrolide resistance in S. pneumoniae has been attributed to an alteration in the L4 and L22 proteins of the 50S subunit [61][62]. Oxazolidinones bind to the 50S subunit but have a more complex set of interactions associated with their mechanism of action [63]. The translocation of peptidyl-tRNA from the A site to the P site is hindered by this class of antibiotics, but enterococci have been documented to have an altered the P site through the substitution of U in place of G in the peptidyl transferase region (position 2576) of the 23S rRNA, thus resulting in a lowered binding affinity in the 50S subunit for this class of antibiotics [64][65][66]. Mutations more closely associated with the A site have been found in E. coli at positions 2032 and 2447 which confer resistance to the oxazolidinone drug linezolid [67].
The 30S ribosomal unit is the target of tetracycline and of aminoglycosides, which function by preventing the decoding of mRNA [68]. Mutations of the gene encoding 16S rRNA confer resistance to this class of antimicrobials [69]. Suzuki and colleagues discovered that substitutions at positions 1400, 1401, and 1483 led to kanamycin resistance in clinical isolates of Mycobacterium, and further strengthened the claim that these changes led to resistance by identifying their absence in kanamycin-sensitive Mycobacterium isolates [70]. Position 1400 was the most common substitution, featuring an A to G change [70]. The same A to G substitution at position 1408 led to high resistance against amikacin, kanamycin, gentamicin, tobramycin, and neomycin in clinical isolates of Mycobacterium abscessus [71].
Active Efflux Pumps of Antimicrobial Agents
In
Bac
aste
s where intact antiria as microbial
pathogens are causative agents
enterof life-threatening infectious diseases [1]. Such pathogenic bacteria
l cells and drug targets are freely accessibleproduce alarming numbers in terms of morbidity and mortality outcomes [2,
3]. activOne
drug efflux systems can come into play. In this section, we will focus on well-studied antimicrobial transpcrucial avenue towards bacterial pathogenesis involves the reduction in the therapeutic effects of antibacterial chemotherapy [4,5]. Thro
rughout
ers, as they make good model systems for study and resistance modulation. B their evolutionary history, bacteria
that are l pathogen
ic frequently make use of integral membrane proteins that function as transporters have developed various means of resisting the inhibitory and bactericidal consequences of antimicrobial agents [
964]. Such
ba
cterial transport proteins serve to actively ntimicrobial resistance systems involve the engagement of bacterial molecular and cellular-based machinery [6]. Inte
xpor
t structurally distinctivestingly, the selection of a bacterial variant with resistance to a single antimicrobial agent
s from the cytoplasm, where drug targets reside, to the extracellular milieu, where their molecular targets are lacking frequently manifests the emergence of a multidrug resistance characteristic in the new mutant [
977].
EffNewl
ux pumps are present in all y emerged bacteria
and are integral parts of l pathogens with resistance to multiple antibacterial
physiology, being involved in diverse functions such as the expulsion of toxic products of magents can result in compromised efficacy in the treatment of infection [3,8].
Me
tcha
bolism, and maintenance of homeostasis. However, antibiotics as incidental substrates of efflux pumps have resulted in them being viewed largely as nisms of antimicrobial resistance include the active export systems within the membranes of bacteria
l mechanisms, prevention of antimicrobial
resistance. In clinically importantentrance into cells of pathogenic bacteria,
such as MDR Mycobacterium tuberculosis, enzymatic destruction of antim
ethic
illin-resistant Staphylococcus aureusrobial agents,
Klebsiella pneumoniae, a production
d Pseudomonas aeruginosa, e of
thick biof
lux pumps have critical roles in ensuringilms, modified targets of antimicrobials, and bacterial s
urvival and evolution into resistant strainsites of action that are protected from antimicrobials, () [2,4].
TFurthe
sermore, multidrug-resistant bacteria
l multidrug efflux pump systems are energetically driven by ATP hydrolysis, called primary active transport [98], a have developed mechanisms that confer the DNA transfer of genetic determinants of resistance to pathogenic species in the clinical setting, the food production ind
bustry
electrochemical ion gradients or ion motive force, the human gut, and in agriculture [9].
Figure 1. Bacterial mechanisms of resistance to antimicrobial agents. The common mechanisms of antibiotic resistance in bacteria are enzymatic hydrolysis (1), enzymatic modifications of antibiotics by group transfer and redox process (2), modifications of antibiotic targets (3), reduced permeability to antibiotics by modifications of porins (4), and active extrusion of antibiotics by membrane efflux pumps (5).
Thus,
callne
d secondary active transport [99,100]. Aw strategies for the c
ti
ve transport ofrcumvention of bacterial resistance to antimicrobial agents
representsare desired [10]. aIn
essential resistance mechanism in bacterial pathogens. Aorder to discover novel approaches to address multiple
structurally distinct antiantimicrobial resistances in these microbial
agents with disparate modes of action are exported to the extrapathogens, however, it is necessary to attain a clear understanding of these resistance systems at the molecular and cellular
milieu, their growth inhibitory properties towardslevels. For young and new investigators, here, we consider an introductory overview of each of these disparate bacteria
are diminished,l resistance mechanisms here.
2. Enzyme-Based Antimicrobial-Inactivation Systems
Along the tif not wholly circumvented.
Dmeline of antibiotic discovery and introdu
rcti
ng the primary active transporton, several enzymatic mechanisms of anti
microbial agents, bacteria exploit the biological energy stored in the form of intact adenosine triphosphate (ATP) to export drugs against the drug concentration gradient by performing ATP hydrolysis [25].biotic inactivation were also discovered. Although very few novel mechanisms of antibiotic resistance have been reported in recent times, several new Duvari
ng the export of antibacterial agents fromants of known enzymes that endow bacteria
l cells, ATP is hydrolyzed in order to energize the drug translocation through the transporter in an outward direction across the membrane. Thus, as the transporter substrate actively with resistance to newly introduced drugs have emerged, suggesting that the bacterial response to new antibiotics or the modified versions of existing antibiotics is swift. The enzymatic mechanisms of antibiotic resistance include hydrolysis, group transfer, and redox processes [4]. In accumulate
s outside the cell, drugrms of diversity, evolution, and spread, antibiotic resistance
is conferred uponenzymes contribute remarkably to the bacterial
pathogen [98]ability to overcome antibiotic pressure.
OnThe β-lactamase
of the best-studied of these primary active drug efflux systems in bacteria is the ATP-binding cassette (ABC) efflux pump family [101,102]s are the oldest known and the most diverse antibiotic degrading enzymes that cleave the β-lactam ring of the penicillin group of antibiotics and render them ineffective. The
ABC tfir
ansporter superfamily represents one of the most abundant protein families known across all taxa of living organisms [103]. One of st such β-lactamase was discovered soon after the first antibiotic penicillin was in clinical use. Scientific evidence suggests the
fexi
rst of the bacterial ABC efflux pump structures to be determined was Sav1866, from the pathogen S. aureus [104] (Figure 5). Strustence of β-lactamases before penicillin was clinic
turally
speaking, the Sav1866 drug efflux pumps consist of two chief transmembrane domains (TMDs) and two nucleotide-binding domains (NBDs)employed, emphasizing that the production of antimicrobial compounds and the mechanisms to endure them occur in parallel in the environment [
10411].
DuBacteri
ng translocation and efflux of the antimicrobial agent across the bacterial membrane, a conformational change occursa that produce antibiotics apparently require mechanisms to overcome the lethal effects of the compounds, and these are in the
TMDs in order to accommodate substrate binding and transport [105]. Meform of concurrent production of degradative enzymes, mutations in targets of antibiotics, or a
nwhcti
le, as the antimicrobial agent is pumped to the outside of S. aureus ve extrusion of antibiotics from the cell so that the antibiotic-producing cell
s, ATP is
hydrolyzed to adenosine diphosphate (ADP) in the interior of the cell by the NBDs, which harbor ATPase activities [104,105].
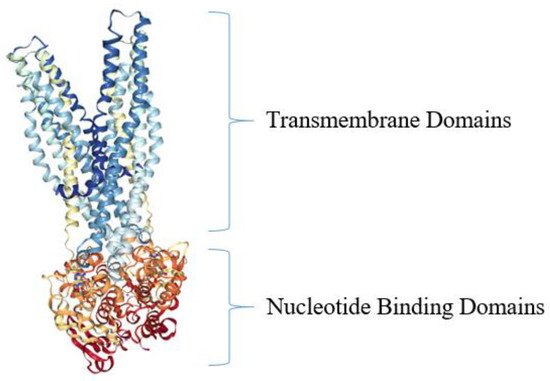
Figure 5. Crystal structure of bacterial ABC efflux pump from S. aureus. The top portion of the ABC transporter Sav1866 is depicted in blue and light blue and represents the two TMDs (sometimes called membrane-spanning domains, MSDs) of the protein, while the orange and red colors depict the two NBDs [104]. The model structure was generated using NGL Viewer [106] of the PDB [107] entries 2HYD and 2ONJ, as reported [104,108].
Tprotected. However, the selection pressure created due to the extensive use of antibiotics in humans and animals propagated the
ABC group of efflux pumps promptresistant clones of bacteria
l recalcitrance to in clinical
ly relevant drugs in Mycobacterium tuberculosis, Acinetobacter baumannii, Streptococcus pneumoniae, Staphylococcus aureus, and food production environme
nt
c. MsrA, widely distributed in Gram-positive and -negative orgs. In due course of time, genetic exchange mechanisms
, is responsible for macrolide facilitated the wider dissemination of resistance
[109].traits Ain
erythromycin inducible MsrA homolog efflux pump, Mel, mediates macrolide resistance in Streptococcus pneumoniae bacterial communities. The introduction of more antibiotics, newer as well modified, augment
oge
ther with MefE [110]. Higher exd the pr
ocess
ion of ABC efflux pumps Rv1217c, and Rv1218c resulted in increased MIC of rifampicin, whil of evolution and spread of resistance mechanisms. Since the
overexpression of Rv1218c increased the MIC of isoniazid [111].majority of the antibiotics introduced in the last two decades are In S. pneumoniae,mostly the
ABCmodified efflux pumps PatA and PatB confer resistance to clinically relevant drugs such as the fluoroquinolones and are overexpressed in clinical isolatesversions of existing antibiotics belonging to the same classes (e.g., β-lactams), a few mutations in the enzymes could render bacteria quickly resistant to them [
11211].
The
Mβ-lac
B efflux pump of E. coli itams
con
e of the few well-studied efflux proteins of the ABC superfamily, which confers appreciable levels of resistance to macrolides [113]. This pstitute the largest group of clinically used antibiotics, comprising of penicillins, cephalosporins of differ
ote
in, together with its outer membrane protein MacA, has been shown to have a crucial role in the virulnt generations, monobactams, and carbapenems, all of which are characterized by the presence of
E. coli. I3-carbon
Salmonella Typhimurium,
MacABC 1-ni
s necessary for host colonization, and it helps the bacterium to overcome the lethal oxidative stress induced by the reactive oxygen species (ROS) and aids in its survival inside macrophages [114].Strogen containing β-lactam ring. The β-lactam antibiotics inhibit the bacterial proteins known as penicillin-binding proteins (PBPs), which pe
crfo
ndary active transporters also confer bacterial resistance to manyrm the critical role of peptide cross-linking during peptidoglycan cell wall biosynthesis. The structura
lly distinctivel mimicry of the d-Ala-d-Ala ant
imicrobial agents [115,116].erminal fragment of Thcro
ughout the last 30 years, these active antimicrobial effluxss-linking peptide by β-lactams facilitates competitive inhibition of PBPs [12], pumpwhich systems have been categorized into several large superfamilies of relatstops the cell wall synthesis leading to bacterial cell lysis and death [13]. Howe
dver, proteins based on similarities in amino acid sequences, structures, and modes of energization [117,118]bacteria gain resistance to lactam antibiotics by modifying their PBPs, which are no longer susceptible to binding by the antibiotic.
CuAlter
rently, these superfamilies are denoted as follows: the major facilitator superfamily (MFS) [119]; natively, bacteria produce powerful lactamases that degrade antibiotics before they can bind with the PBPs. Since their discovery in the
drug/me
tabolite transporter (DMT) superarly 1940s, the family
, which now harbors the small multidrug resistance (SMR) family [120 of β-lactamases has grown seamlessly,
121]; with
e multidrug and toxic compound extrusion (MA more than 300 enzymes identified globally [14,15]. T
E)he family, which has been included within the larger multidrug/oligosaccharidyl-lipid/polysaccharide (MOP) superfamily of transportersearly β-lactamases were penicillinase enzymes that degraded penicillin, which started appearing rapidly in clinical bacteria [
12216,
12317]
;. the proteobacterial antimicrobial compound efflux (PACE) transporter superfamily [124]; aThe introduction of modified, semisyn
d the
resistance-nodulation-cell division (RND) superfamily [125]. Stic penicillins such as methicillin, ampicillin, and amoxicillin resulted in the gradual appe
veara
l well-studied families of bacterial solutnce of β-lactamases capable of degrading them. The first plasmid-borne trans
porter systems are shown in Figure 6.
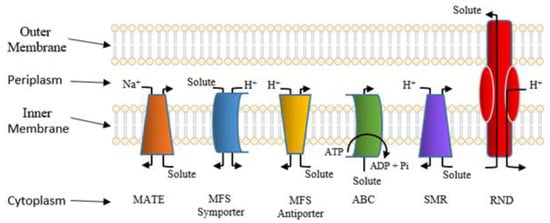
Figure 6. Classes of well-studied bacterial solute transporters. The bacterial outer and inner (cytoplasmic) membranes are shown. Also depicted are the cytoplasmic and periplasmic spaces. Pi denotes phosphate, and Na+ and H+ denote sodium and proton, respectively. This figure kindly provided courtesy of Ann F. Varela.
ferrable β-lactamase was TEM-1, followed by TEM
-2 an
y members ofd SHV-1 enzymes [18,19]. TEM is the
MFSmost of bacterial efflux pumps confercommon mechanism of ampicillin resistance
to multiple antimicrobial agents and are considered essential molecular targets for resistance modulation in order to circumvent resistance and restore the therapeutic efficacy of compromised agents [126,127].compared to less prevalent SHV-1, although both have the same affinity for this antibiotic. TEM and SHV share 60% amino acid similarity between them and are inhibited by clavulanic acid, tazobactam, and sulbactam. The discovery of cephalosporin C in the early 1960s heralded an era of synthetic cephalosporins, which was thought to Thfe
protein snd off β-lactamases. Structur
es for several bacterial antimicrobial efflux pumps belonging to the MFS have been elucidatedally, cephalosporins have their β-lactam ring fused to a six-membered dihydrothiazine ring compared to penicillins in which the β-lactam is fused with a five-membered thiazolidine ring [
12820].
ISubsequen
general, the MFStly, carbapenem and monobactam groups of β-lactam antibiotics with structur
es harbor 12 or 14 α-helical transmembrane segments, two seemingly symmetrical bundles, each belonging to either the N- or C-terminal ends, the so-called MFS fold consisting of adjacent triplet α-helices, and functional highly conserved amino acid sequence motifally variant lactam rings were discovered from natural sources and formed the basis for the synthesis of similar compounds with modifications. However, the enzymes extended-spectrum β-lactamases (ESBLs) that could hydrolyze a wide range of cephalosporins emerged from TEM and SHV lactamases by point mutations [
128,12918].
RecentESBLs hydroly
, protein structure studies of the MdfA multidrug efflux pump from E. coli ze a broad spectrum of cephalosporins, including first, second, third-generation cephalosporins
howed bound substrates, such as chloramphenicol [130] (Figure 7)and aztreonam, but not cephamycins and carbapenems, and
are inhibit
ors,ed by clavulanic acid [
13018,
13121]
,. plus a crystal structure composed of a periplasmic-facing conformation suggesting a functional role for the highly conserved antiporter motif C As a consequence of mutations and the expansion of the substrate range, ESBLs have a lesser affinity for classical β-lactams compared to their ancestral β-lactamases. Subsequen
ce in conducting substrate translocation through the antimicrobial pumps [132,133,134].tly, CTX-M type ESBLs with high affinity for cefotaxime emerged independent of TEM and S
HV lact
udies likeamases, and these
will undoubtedly play crucial rolesupposedly evolved from β-lactamases of Kluyvera s
pp. [22]. inOver the
evaluation of the physiological mechanisms for antimicrobial efflux acryears, CTX-M has overtaken other ESBLs in terms of number and global distribution, with more than 230 types identified to date. sho
sws the
membrane and their exploittimeline of the evolution of β-lactamases in relation
for the development of efflux pump inhibition [135]to the introduction of β-lactam antibiotics for clinical use.
Figure 72. CrystaEvol
structure of E. coli MdfAution of β-lactam
ultidrug efflux pump from the MFS. The MdfA transporter is complexed to one of its substrates, chloramphenicol (ball and stick structure). Ribbons of different colors represent the transmembrane helicesases. Within five decades of discovering the first penicillin-degrading enzyme, β-lactamases capable of hydrolyzing most β-lactam antibiotics, and resistance to inhibitors have emerged. The
loops between the transmembrane domains were removed for clarity. The model of the MdfA structure was generated using NGL Viewer [106]ability to tolerate a broad spectrum of β-lactams and inhibitor combinations is bolstered by from the
Protein Database, PDB [107], presence of multiple β-lactamase-encoding gen
tryes 4ZOW from Heng et alin a single pathogen.
[130].
Som
The
initial effo
f the clinically relevant and intensely studied MFS efflux pumps belong to Staphylococcus aureusrts to classify β-lactamases were based on their functional characteristics such as the substrate-inhibitor profiles,
protein
cluding NorA, NorB, NorC molecular weight, isoelectric point, etc. [12,14,
23]. A Qasec
A, QacB, TetA(K), LmrS, and MsrA [136]. Thond approach employed amino acid se
sque
efflux pumps directly or indirectly contribute to the ability of Staphylococcus aureus nce similarities and enzymatic activities to classify β-lactamases into
tfo
lerate antibiotics, such as by decreasing intracellular concentration of antibiotics, which allows bacteur main groups, of which groups A, C, and D are serine β-lactamases, while class B is composed of metallo β-lactamases that require active site zinc ion(s) for their hydrolytic activities [12,24]. Gr
iaoup to survive longer in the presence of antibiotics and developA enzymes form the largest group of lactamases comprising some of the critical resistance
through other mechanisms involving gene mutations, overexpression of porins, etc. Inenzymes such as TEM, SHV, and CTX-M type of β-lactamases. Other important ESBLs include the carbapenem hydrolyzing KPC type ESBLs originally reported from S.Klebsiella aurpneumoniaeus,
which the NorA efflux pump promotes the development of ciprofloxacin resistance directly or by positively contributing to the fitness advantaghave an expanded substrate spectrum encompassing the cephalosporins and carbapenems but susceptible to inhibition by clavulanates and boronic acid [23,25]. The
pchro
vided by topoisomerase gene mutations [137]. Tmosomally encoded AmpC (class C) cephalosporinases described early in the
timel
evated levels of norA ine of the discovery of β-lactamase
xprs have
ssion potentiate ciprofloxacin resistance, although this phen no homology with penicillinases and thus constitute a distinct group of enzymes [26,27]. Co
mm
enon is highly variable across clinical staphylococcal strains [137]. Ionly found in Enterobacteriaceae, AmpC enzymes are inducible and are produced at low basal levels, and preferentially hydrolyze cephalosporins in
hcludi
bition of the NorA efflux pump with a clinically approved drug nilotinib diminished the biofilm formation by S. aureus,ng cefoxitin but not cefepime. These are generally resistant to inhibition by clavulanic acid, sulbactam, or tazobactam. The metallo-β-lactamases or MBLs belonging to cla
nd this drug can potentiate ciprofloxacinss B have vigorous hydrolytic activities against carbapenems and are also activ
ity in clinical settinge against a range of cephalosporins [
13828,29].
ObIn 2009, a new v
iously, efflux pumps are key components of complexariant New Delhi Metallo-β-lactamase (NDM), emerged, and since then, it has been reported from all over the world [29]. NDM c
ionfer
cuits involvings resistance to all β-lactam antibiotic
resistance, persistences except aztreonam, and
virulthe plasmid carrying blaNDM gen
ce
[139].
Witharbors the discovery of the SMR family and its subsequent incorporaresistance markers for several other antibiotics. VIM and IMP are other important class B carbapenemases commonly encountered in Enterobacteriaceae.
The OXA t
iype enzymes belon
inging to the
larger DMT superfamily arose the elucidation of a low-resolution crystal structure for the DMT-baseClass D lactamase group were originally discovered as plasmid-encoded oxacillin hydrolyzing enzymes in lactose non-fermenting bacteria such as Pseudomonas, Acinetobacter, and
Shewanella, an
d lat
imicrobial efflux pump, called EmrEer in Enterobacteriaceae through plasmid exchange [30,
31]. wTh
ich has been an effective model system for antimicrobial transport [121,140,141]ese enzymes are poorly inhibited by lactamase inhibitors such as clavulanic acid.
WAlthough
ile the structural nature of EmrE has been controversial in terms of the monomer orientation for its dimer [141,142], mole OXA lactamases have a narrow substrate range composed of penicillins, cloxacillin, and oxacillin, the enzymes evolved to hydrolyze extended-spectrum cephalosporins and c
ular
dynamics simulbapenems through point mutations
, biochemical, and
physiological studies pertaining to tthese abilities vary among different OXA types [28,32].
The
β-lactamas
tructure-function relationships ane mediated antimicrobial resistance is widespread among ESKAPE (Enterococcus, S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa, and
E. coli) egroup of
flux inhibition have shed new light on its substrate translocation mechanism organisms, infections with which are usually associated with a significantly higher economic burden and highest risk of mortalities [
14333,
144,145,146,14734].
The
cWor
ystal structure of the RND transporter AcrB from E. coli, fld Health Organization (WHO) has recognized carbapenem-resi
rst
reported in 2002 [148],ant Enterobacteriaceae (CRE) as a cons
ists of a trimer [149,150]. Terious global health scourge for wh
e Aic
rB trimer component is known to reside wih the development of new antimicrobials is critically needed [35].
Enzymat
hi
n the inner membrane of Gram-negative bacteria [151]c hydrolysis is also a common mechanism of resistance against macrolides, rifampicin, and fosfomycin.
IMan
one mechanistic model for antimicrobial transport, the AcrB is thought to rotate in a manner akin to a peristaltic pump in whichy Enterobacteriaceae members produce plasmid-encoded esterases EreA and EreB that hydrolyze the macrolactone ring of 14- and 15-membered macrolides such as erythromycin A, clarithromycin, and azithromycin [36,37]. tThe
pump repeatedly cycles between extrusion, access, and binding steps [152,153].structurally altered macrolide antibiotic will no longer be able to bind to its preferred target site in Furthe
rmore, ribosome [38].
Anothe
Acr
B efflux pump has been demonstrated to as important mechanism of enzymatic degradation is associated with the manganese ion (Mn2+)-dependent, chromos
eom
ble into a tripartite multi-complex assembly with a periplasmic-located protein, Acrally-encoded FosX that uses water to cleave the epoxide ring of fosfomycin. Other fosfomycin modifying metalloenzymes include FosA, FosB, and two epoxide kinases FomA and FomB [39]. FosA
, is a Mn2+ and
aK+-dependen
t outer-membrane protein, TolC [154].glutathione-S-transferase, while FosB This
a Mg2+ t
hiol-S-tr
ipartite antimicrobial drug efflux system has been found in a varietyansferase. The mechanism involves adding glutathione or thiol groups to the oxirane ring of fosfomycin resulting in an inactive drug [40]. FomA and Fo
f life-mB kinases utilize ATP and Mn2+ ions t
o ph
reatening bacosphorylate the oxirane ring of fosfomycin [39].
Tet
er
ial pathogens and confers resistance to multiple acyclines are in use for over 70 years as widely used antibiotics in human and animal medicine [41]. Tetrac
yclin
ically relevant antibacterial agentse is broken down by a monooxygenase enzyme Tet(X), which is oxygen- and FAD-dependent [
15542]
. T
he
bacterial RND tripartite multidrug efflux pump systems from E. coli ct(X) monohydroxylates break down tetracyclines at position 11a, followed by non-enzymatic degradation. Similarly, enzymatic mono
xygen
sistsation of th
ree main domains constituting a tripartite structure. The top thirde naphthyl group of rifamycin antibiotics by monooxygenases (Rox) inactivate them by leading to the linearization of the
structure denotes the outer membrane-associated channel, TolC; naphthoquinone or naphthohydroquinone ring [43].
Enzymat
heic m
iddle section includes the periplasmic-associated domain, AcrA, and the third section is constituted by AcrB, an extensively studied member of the RND superfamilyodification of antibiotics by the transfer of functional groups, such as acyl, glycosyl, ribosyl, nucleotidyl, phosphoryl, or thiol groups, confers resistance to a range of antibiotics, including aminoglycosides, rifamycins, macrolides, epoxides, and chloramphenicol [
15044].
I The amin
oglycoside general, these distinctive families of antimicrobimodifying enzymes (AME) responsible for resistance to different aminoglycoside antibiotics include N-a
cetyl
transporter systems transferases (AAC), O-adenyltrans
fer
ve tases (ANT), and O-pho
cspho
nfer bacterial pathogens enhanced capabilities to survive antimicrobial stress [136]. Aptransferases (APH). These enzymes catalyze the modification of various hydroxyl or the a
rt frmino
m AcrB-TolC, some groups of the
extensively studied, clinically relevant RND efflux pumps are MexB, MexF, and MexY of Pseudomonas aeruginosa,aminoglycosides resulting in their inability to bind to Adthe
B of Acinetobacter baumannii,ir 30S ribosomal Cmtarge
B of Campylobacter jejuni,ts [45]. Simila
nd MtrD of Neisseria gonorrhoeae rly, in Gram-negative bacteria
, [156].a In Bacteroides fragilis cpl
asmi
nical isolates, bmeB d-encoded ADP-ribosyltransferase
fflux (Arr-2) is pump overexpression coupled with GyrA point mutations contribute tocommonly responsible for rifampin resistance [46]. Simila
clinical level of resistance to fluoroquinolone and β-lactamrly, chloramphenicol is modified by acetyl-CoA-dependent acetylation of its 3-hydroxyl group by chloramphenicol acetyltransferase (CAT) enzymes [
15747].
AThe recent study suggests that the AcrAB efflux pump has a role in the initial stages of modified antibiotic does not bind to its target site, the 50S subunit of ribosomes. CATs are widely distributed among Gram-positive and -negative bacteria
l transition from transient antibiotic resistance to permanent resistance. The lower expression of DN and show little amino acid sequence similarities, with only 25 amino acid residues conserved among all CAT variants [47].
3. Alteration of Antimicrobial Targets
A
s repair gene mutS bacterial enzymes menti
on
acrAB ed above
alter
expressing strains contributes to higher frequencies of spontaneous mutations and hence higher probabilities of drug structures, the drug targets may likewise be altered, preventing drug binding and, thus, conferring resistance
development [158]. The. Antimicr
efo
re, the presence of an efflux pump and its expression level cannot be viewed in isolation but should be correlated with other mechanisms of resistance that might act in synergy with efflux pumps. Consequently, these drug transport systems represent desirablebial targets play vital roles in microbial growth or survival and, thus, serve as potentially useful targets for mitigating infection. In addition, these targets must differ or be completely absent from humans or the animal species being treated with an antimicrobial to allow for a selective mode of action. A classic example of such a target
s for inhibitors [159] is pepti
doglycan
order to circumvent resistance and restore the therapeutic efficacy of multidrug-resistant. Peptidoglycan is essential to the growth and survival of many bacterial
pathogens [10,126,127,128,136,160].species and has a Tche
refore, molecular studies of transporter structures and efflux mechanisms will undoubtedly continue to be relevant in the foreseeable future [161].Umical structure that is not present in the mammalian hosts they infect. This allows for the targeting of enzymes responsible for the syn
fort
unately, fundamental knowledge of the molecular mechanisms for multidrug transport is lacking. For example, we still know little about the modes for tying together energetic systems versushesis and assembly of peptidoglycan. The function of proteins associated with these target sites makes it non-viable for a bacterium to evolve resistance by removing these proteins. However, mutations that allow for continued functionality while reducing the ability of an antimicrobial
translocation across the membrane. Further, we do not yet understand howagent to bind them at the target site have been a veritable regularity in the arms race between antimicrobial
transporters dictate multiple substrate transport while preventing the passage of unwanted substrates or leakages of relatively smaller ionsubstances and antimicrobial-resistant bacteria. In addition to peptidoglycan, alteration in target sites has been attributed to ribosomes, nucleic acid enzymes, and lipopolysaccharides [48].
As
, like sodium ions or protons. For many if not alldiscussed previously in this review, peptidoglycan inhibition by glycopeptides involves the binding of the
se peptidyl-d-alan
timicrobiyl-d-al
anine t
ransporters we do not yet have a clear picture of the nature of the structural configuerminus of peptidoglycan precursors. This binding prevents integration
s assumed during each via the transglycosylase activity of the
specific steps of theirse precursors into the cell wall [49], tra
nsport cycles. Ins shown in .
Figure 3. Penicillin and penicillin-binding protein of the bacterial cell wall. (1) The peptidoglycan layer of a bacterial cell wall harbors the repeating moieties of N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM). The NAM subunits bind short variable peptide chains, usually l-Ala and two distal d-Ala residues. (2) The PBP cross-links the peptide side chain, releasing a free Ala. (3) Upon cross-linking, the PBP dissociates from the cell wall. (4) Penicillin binds the PBP active site, affecting its enzyme activity. (5) The β-lactam ring of penicillin is cleaved during its reaction with PBP. Penicillin stays covalently bound PBP, permanently inhibiting the active site. Altered PBPs, such as PBP2a, are unable to accommodate penicillin-binding, preventing cell wall synthesis inhibition [48,49].
PCBs summary, much work remains to be performed before we can clearly understand the physiologe one mechanism for antimicrobial resistance, but the peptidoglycan precursors themselves can undergo alteration, which reduces the affinity of antimicrobial transport both at fundamental and applied levels of investigs without the involvement of enzymatic inactivation.
Future Directions
Ba Suc
terh i
al pathogens are crits the case with Enterococcus faecium and E. faecalis, whic
h ha
lly essential causative agents of severe infectious disease [184].ve been discussed in the literature as developing resistance by As suac
h, much effort has gone into the development of chemotherapy in addressing high morbidity and mortality numbersquiring one of two related gene clusters encoding VanA and VanB [
18550,
18651]. The
rse ge
fore, continued investigation towards the improvements in personne clusters produce a modified terminus that contains d-al
hany
gienl-d-lactate
methodas
, food h opposed to d-alan
dyl-d-al
anin
ge [50]. This a
nd prlte
paration
, hand washing, public sanita leads to glycopeptides having a much lower binding affinity [52]. Thus, t
iohese gen
, and education across all levels will be the focus of intense intereste clusters, found on transposable elements, have allowed the spread of modified targets in enterococci.
In Sim
edical healthcare and treatment centers, antimicrobial stewardship is still a promising approilarly, there are rarer but related gene clusters that have been shown to modify peptidoglycan precursors, such as those encoding VanD [53], Va
chnE [54], and
Van G [55].
Ribosom
uces, serving th
effort continues to be centered towards further development [187,188].e vital role of protein synthesis, are common to both prokaryotic and eukaryotic organisms but differ quite vastly from one another in Ast
tention will undoubtedly need to be paid towards studies of multidrug resistancructure, making them another suitable candidate for antimicrobial targeting [56]. The
50S ri
n bacteria found in veterinary medicine and agribosomal unit serves as the binding site for macrolide, lincosamide, and streptogramin B [57]. Rec
ual
tural practices to reduce infection transmission and persistence icitrance to these specific antimicrobials is known as MLS(B) type resistance [57], an
d these areas [9].
New it results from a post-transcri
ncenpti
ves to discover new antibacterial agents with novel modes of action are few, and progress on this front is slowonal modification of the 23S rRNA component of the 50S ribosomal subunit that is involved with methylation or dimethylation of key adenine bases in the peptidyl transferase functional domain [
189,19058].
A prMutatio
mising avenue in the battle against multidrug-rns in the 23S rRNA, close to the site of methylation have also been associated with resistan
t pathogens entails the clinical investigation of non-ce to the macrolide group of antibiotic
agents ass in a range of organisms, such as Helicobacter pylori [59] an
td propioni
-bacteria
[60]. Macrol
ide agents, sucresistance in S. pneumoniae h
as
non-steroidal anti-inflammatory agents, anesthetics, and statins [191].been attributed to an alteration in the L4 and L22 proteins of Rthe
cently 50S subunit [61,
62]. Oxa
series of new and well-developed anti-infective strategies forzolidinones bind to the 50S subunit but have a more complex set of interactions associated with their mechanism of action [63]. tThe
circumventranslocation of
multidrug-resistant pathogens were reviewed elsewhere [10].peptidyl-tRNA from the A site to the P site is hindered by Tth
ese and other strategic modes for reducing the conditions that foster the spread of bacterial infections are prime candidates for enhanced efforts of investigation.
8. Concluding Remarks
Bis class of antibiotics, but enterococci have been documented to have an altered the P site through the substitution of U in place of G in the peptidyl transferase region (position 2576) of the 23S rRNA, thus resulting in a
ct lower
ial pathogens that have acquired specific antimicrobied binding affinity in the 50S subunit for this class of antibiotics [64,65,66]. Muta
ltions resistance mechanismsmore closely associated with the A site have
emerged as serious clinicbeen found in E. coli a
l agent
s of infection, causing a public health concern on a worldwide scale positions 2032 and 2447 which confer resistance to the oxazolidinone drug linezolid [67].
The 30S
uch cellular mechanisms of antimicrobial resistance include multidrug efflux pumps, enzymatic drug degradation, biofilm formation, drug target modific ribosomal unit is the target of tetracycline and of aminoglycosides, which function by preventing the decoding of mRNA [68]. Mutation
,s and target protection. Many genetic determinants for bacterialof the gene encoding 16S rRNA confer resistance to this class of antimicrobial
s [69]. resSuzuki
stance are transferable to unrelated species, having evolved new means of movement through human populations. To and colleagues discovered that substitutions at positions 1400, 1401, and 1483 led to kanamycin resistance in clinical isolates of Mycobacterium, reand
uce the conditions further strengthened the claim that
foster the emergence and spread of clinical infections new strategies have been considerthese changes led to resistance by identifying their absence in kanamycin-sensitive Mycobacterium isolate
ds [70].
Future dPosi
rections include the development of new chemotherapeutics, such as those with noveltion 1400 was the most common substitution, featuring an A to G change [70]. cThe
llular targets, the continuation of public health practices, education, clinical antimicrobial stewardship same A to G substitution at position 1408 led to high resistance against amikacin, kanamycin, gentamicin, tobramycin, and
continued molecular investigation of resistance mechanismsneomycin in clinical isolates of Mycobacterium abscessus [71].