Plant polyphenols, such as flavonoids, chalcones, triterpenes, and proanthocyanidins, have cardioprotective, antiapoptotic, and anti-ischemic properties, which cause a decrease in the infarct size, arrhythmia score, and improvement in cardiac stunning primarily via the release of NO, the activation of the Nrf2 pathway and endogenous antioxidant defense system
[77][88]. Resveratrol, the polyphenolic compound present in red grapes and wine, ameliorates cardiac dysfunction induced by MI/R by activating the Nrf2/ARE pathway and increases the expression of Nrf2 by inducing SIRT1 or inhibiting GSK3β, thus alleviating myocardial oxidative stress, and thereby improving IRI
[78][79][89,90]. It was suggested that through the activation of PI3K/Akt/p38 MAPK, H
2O
2 preconditioning alleviate the caspase-3 activity, increases the expression of Bcl2, and leads to the translocation of Nrf2 into the nucleus, which selectively increases the expression and activity of antioxidant enzymes to prevent MIRI-induced apoptosis
[65][76]. Triptolide, the bioactive component present in
Tripterygium wilfordii Hook F, reportedly reduces I/R-induced myocardial infarction, inflammation, oxidative stress and improves the cardiac function in rats via its effect on the Nrf2/HO-1 defense pathway
[80][91]. Aloin, the major bioactive anthraquinone of the Aloe species, also diminishes I/R-induced oxidative stress injury and inflammatory response in cardiomyocytes by activating the Nrf2-HO-1 signaling
[81][92]. Recent studies indicated that garlic (
Allium sativum) and its constituents, such as Allicin and Diallyl sulfide, were reported to activate Nrf2 and increase HO-1 and NQO1 expression through ERK/p38 signaling pathway activation, thus playing a protective role in the adaptation of diabetic cardiomyopathy
[82][93]. Another polyphenolic compound, luteolin, contained in vegetables, was shown to protect the diabetic heart against IRI by enhancing eNOS-mediated S-nitrosylation of Keap1, with subsequent upregulation of Nrf2 and the Nrf2-related antioxidative signaling pathway
[83][94]. New curcumin analog, 14p, decreased oxidative stress and reduced MIRI via increasing Nrf2 expression
[72][83]. Kaempferide (3,5,7-trihydroxy-4-methoxy flavone) inhibits Nrf2 and cleaved caspase-3 signaling pathways through a PI3K/Akt/GSK 3β-dependent mechanism and attenuates I/R-induced myocardial injury
[84][95] (). Simultaneously, Butin, a plant dietary flavonoid, protects against I/R-induced ROS-mediated apoptosis by upregulating the AMPK/Akt/GSK-3β pathway and activates Nrf2-regulated antioxidant enzymes in diabetic cardiomyocytes exposed to I/R
[85][96]. Crocin, the active component of saffron, suppresses I/R-induced cardiomyocyte apoptosis via ER stress inhibition by modulating the miR-34a/Sirt1/Nrf2 signaling pathway
[86][97]. Hyperoside, a flavonoid isolated from Rhododendron ponticum L., was suggested to have a protective effect on cardiac IRI by inhibiting ER stress and activating the Nrf2 signaling pathway in an ischemia/reperfusion animal model
[87][98]. Soybean isoflavones are shown to activate Nrf2-mediated antioxidant responses in a dose-dependent way and alleviate ischemic cardiomyopathy
[88][99].
3.2. Hepatic IRI
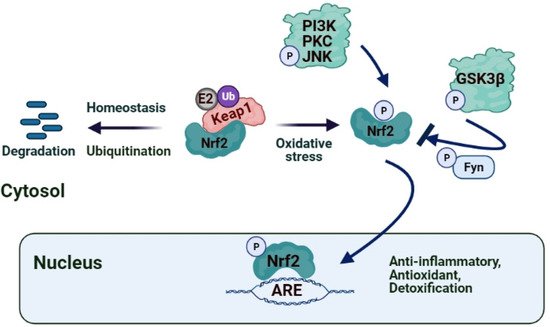
The role of Nrf2 in hepatic I/R was identified by several studies showing that the Keap1–Nrf2 complex may alleviate oxidative injury
[89][100]. Nfr2 was proposed as a fundamental transcription factor involved in diminishing hepatic IRI since the interaction between Keap1 and Nrf2 is disrupted in I/R induced stress conditions, and Nrf2 translocates into the nucleus promoting the transcription of cytoprotective and antioxidant genes such as heme oxygenase 1 (HO-1)
[59][70]. The triterpenoid CDDO-imidazoline (CDDO-Im), a synthetic oleanane triterpenoid and potent activator of the Nrf2 pathway, was found to ameliorate liver IRI in mice by inducing the expression of Nrf2 target gene HO-1, which lead to enhanced autophagy in hepatocytes, and to reduced ROS production and inflammatory responses
[58][69]. Ibrahim et al.
[90][101] studied the protective effects of dimethyl fumarate, which is an FDA approved immunomodulatory drug that induces the anti-inflammatory stress protein HO-1, and curcumin, a polyphenol found in the rhizome of the herb
Curcuma longa, against acute hepatic injury induced by I/R as a combination therapy focusing on the Nrf2/HO-l signaling pathway. They concluded that the combination of these two drugs has a synergistic hepatoprotective effect against hepatic-induced I/R via activating Nrf2/HO-1 pathways. 15-Deoxy-Δ12,14-prostaglandin J2 has been proposed to alleviate hepatic IRI in mice via inducing the Nrf2 pathway, which leads to inhibition of ROS generation, apoptosis, and autophagy
[91][102]. Propofol post-conditioning was postulated to ameliorate hepatic IRI by reducing ROS levels through the enhanced expression of Nrf2. Pretreatment with Sulforaphane, an Nrf2 agonist, reduced apoptosis and oncosis via activating the Nrf2/ ARE pathway, ameliorated oxidative stress, and attenuated hepatic IRI
[92][103]. Telluric acid, a tellurium-based compound, was reported to ameliorate hepatic I/R-induced injury in rats by affecting TLR4, Nrf2, and PI3K/Akt signaling pathways
[93][104]. The transmembrane G protein-coupled bile acid receptor (TGR5) plays a vital role in regulating energy homeostasis, glucose metabolism and possesses protective activity for inflammation by suppressing the NF-κB signaling pathway. Zhuang et al.
[94][105] recently demonstrated that TGR5 reduced hepatocellular apoptosis and inhibited inflammatory response by regulating Keap1-Nrf2 signaling pathways, suggesting that the TGR5 molecule may appear as a novel and potential therapeutic target for preventing hepatic IRI. The IRG1/Itaconate pathway was recently suggested to activate Nrf2 in Hepatocytes and improve I/R induced hepatic injury. 4-IO, a new itaconate derivative, was found to increase the expression and nuclear translocation of Nrf2 and up-regulation of its downstream protective pathways (HO-1 and NQO1) during hepatic injury both in mouse and human hepatocytes
[60][71].
Brahma-related gene 1 (Brg1), a catalytic subunit of SWI2/SNF2-like chromatin remodeling complexes, is suggested to activate the Nrf2/HO-1 pathway during hepatic I/R. Recently, Ge et al.
[95][106] showed that hepatic Brg1 overexpression during reperfusion enhanced Nrf2-mediated inducible expression of HO-1 during hepatic I/R, causing an increase in antioxidant ability and the recovery of liver IRI. Perindopril, an ACE inhibitor, was recently presented to be beneficial in hepatic IRI by suppressing hepatic oxidative and inflammatory stresses through the modulation of NF-κB-p65/TLR-4, JAK1/STAT-3, Nrf-2, and PI3K/Akt/mTOR signaling pathways
[96][107]. Sevoflurane, an inhalation anesthetic widely used in clinical practice, has been observed to induce oxidative stress and affect cognitive functions
[97][108]. More recently, Ma et al.
[98][109] postulated that sevoflurane promoted Nrf2 entry into the nucleus, upregulated the downstream target gene HO-1, activated the Nrf2 gene pathway, and protected the liver from IRI.
Fisetin (3,3′,4′,7-tetrahydroxyflavone), a flavonoid found in various fruits
[99][110]; Gastrodin, a bioactive compound extracted from the traditional Chinese herbal agent
[100][111]; Myricetin, a flavonoid glycoside extracted from the fruits, leaves, and branches of
Myrica cerifera [101][112] and CDDO-Im
[58][69] were reported to alleviate liver damage and oxidative stress in liver I/R through the Nrf2/HO-1 signaling pathway, suggesting that these compounds may be considered as targeted drugs for liver IRI treatment.
3.3. Intestinal IRI
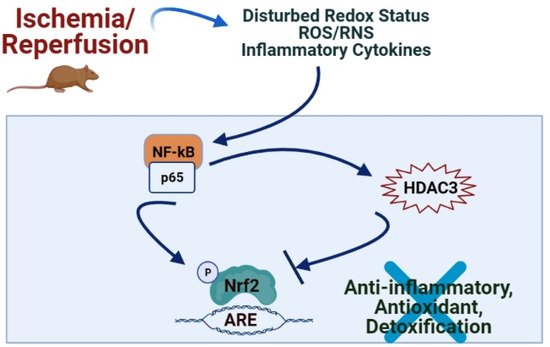
Redox imbalance, nitric oxide, leukocyte adhesion molecule P-selectin, NF-κB, TNF-α, IL-1β, cellular adhesion molecules, and inducible nitric oxide synthase (iNOS) play a central role in intestinal IRI
[7]. Interleukin-1 receptor antagonist (IL-1Ra) possesses a protective effect on intestinal IRI by anti-inflammatory, antioxidative, and antiapoptotic mechanisms via the activation of Nrf2/HO-1 and MAPKs pathways, suggesting it as a new therapeutic drug for the improvement of intestinal IRI
[102][113]. Irisin, a new peptide, promotes heat production, ameliorates obesity, and regulates glucose homeostasis to improve I/R-induced intestinal inflammatory response, reduce oxidative stress, and inhibits apoptosis, which could be associated with the Nrf2 pathway activation
[103][114]. Dimethyl fumarate activates antioxidant pathways, induces Nrf2/HO-1, GSK-3β Wnt/β-catenin signaling pathways, and improves I/R-induced intestinal inflammatory response
[104][115]. Salvianolic acid A ameliorated oxidation inhibited the release of pro-inflammatory cytokines, and alleviated apoptosis in I/R-induced injury through the regulation of Nrf2/ HO-1 pathways
[105][116]. Ginsenoside Rb1, a major active saponin isolated from ginseng, attenuates intestinal ischemia/reperfusion-induced inflammation and oxidative stress via activation of the PI3K/Akt/Nrf2 signaling pathway
[106][117]. Higenamine (1-[(4-hydroxyphenyl) methyl]-1,2,3,4-tetrahydroisoquinoline-6,7-diol) is an active ingredient of
Aconiti Lateralis Radix Praeparata with anti-inflammatory properties, and it has been shown to regulate the Nrf2-HO-1-High mobility group box 1 protein (Hmgb1) axis and attenuate intestinal IRI in mice which appears as a novel therapy to protect intestinal IRI
[107][118].
3.4. Cerebral IRI
Ischemic stroke is a worldwide debilitating clinical disorder and one of the most common causes of death. Reoxygenation of the ischemic brain causes IRI. Excessive ROS production is related to cerebral IRI in stroke. Recent studies are focused on the therapeutic potential of targeting the Nrf2/HO-1 pathway in brain injury after ischemic stroke
[108][119]. 11-Keto-β-boswellic acid, the pentacyclic triterpenoid compound of Boswellia serrata resin, increased Nrf2 HO-1 expression in a middle cerebral artery occlusion model of ischemic injury and protected the brain from oxygen and glucose deprivation-induced oxidative insult
[109][120]. Hu et al.
[110][121] postulated that Panax notoginseng saponins (PNS) activate Nrf2 antioxidant signaling depending on the PI3K/Akt pathway and protect against oxygen-glucose deprivation/reperfusion-induced BBB dysfunction. Z-ligustilide, the main lipophilic component of
Radix Angelica sinensis, reportedly activates the Nrf2/HO-1 pathway, induces Nrf2 nuclear translocation, and upregulates HO-1 expression in a time- and concentration-dependent manner
[111][122]. RS9, a novel Nrf2 activator obtained from bardoxolone methyl (BARD), showed neuroprotective effects against cerebral I/R via targeting NF-κB and Nrf2-ARE pathway and attenuated neuroinflammation
[112][123]. Nomilin, a triterpenoid extracted from citrus fruits, exhibits many biological activities, including immunomodulatory activity. Nomilin ameliorates blood–brain barrier disruption in the middle cerebral artery occlusion rat model, possibly via alleviating the loss of the tight junction proteins ZO-1 and occludin-5 and increasing the expressions of Nrf2 and NQO1
[113][124]. Monomethyl fumarate, a pharmacologically active metabolite of dimethyl fumarate, has been reported to reduce cerebral edema by attenuating oxidative stress and inflammation
[114][125]. Singh et al.
[115][126] recently demonstrated the potential of monomethyl fumarate’s activity as an Nrf2 activator in the peri-infarct cortex. Resveratrol, a very well-known stilbene, has been shown to possess neuroprotective effects primarily by reducing oxidative stress and mitochondrial dysfunction
[116][127]. Pterostilbene from blueberries was reported to exhibit neuroprotective effects via Nrf2 activation
[117][128]. Xu et al.
[118][129] recently compared the neuroprotective effects of pinosylvin, pterostilbene, pinostilbene, and 4-methoxy-trans-stilbene using oxygen and glucose depletion/reperfusion model in PC12 cells. They indicated that pinosylvin has a significant neuroprotective effect by inducing PINK1/Parkin mediated mitophagy and also activating the Nrf2 pathway to ameliorate oxidative stress-induced mitochondrial dysfunction. Further, 2-Cyano-3,12-Dioxooleana-1,9-Dien-28-Oic acid (CDDO), and its derivatives exhibit anti-inflammatory and antioxidant activities. In addition, 6 CDDO-ethyl amide (CDDO-EA) increased HO-1 expression, and primed microglial polarization toward M2 phenotype under inflammatory stimulation in BV2 microglial cells, suggesting that CDDO-EA confers neuroprotection against cerebral ischemic injury
[119][130].
3.5. Renal IRI
Acute kidney injury (AKI) caused by ischemia-reperfusion also constitutes a major clinical issue. RTA-408, a new oleanane triterpenoid compound, induces Nrf2 and downstream antioxidant gene expression, protects the kidney from renal I/R by activating the Nrf2 antioxidant system and increasing the Nrf2 nucleus translocation and subsequently upregulating of downstream antioxidant genes, including GSH biosynthesis
[120][131]. Hyperglycemia-induced oxidative stress is involved in impaired SIRT1/Nrf2/HO-1 signaling and ischemia AKI in diabetes. Melatonin was proposed to ameliorate ischemia AKI in diabetes by improving the SIRT1/Nrf2/HO-1 signaling, suggesting that the SIRT1/Nrf2/HO-1 pathway can be a new target in decreasing the oxidative stress in diabetic IRI
[121][132]. Synthetic triterpenoids such as CDDO (2-cyano-3,12-dioxoolean-1,9-dien-28-oic acid) and its methyl (CDDO-Me, bardoxolone methyl) and imidazolide (CDDO-Im) derivatives have been shown to activate Nrf2-mediated antioxidant activity in several diseases, including chronic kidney disease. CDDO-Im treatment upregulated Nrf2 target gene activation, induced the expressions of cytoprotective genes, including GCLC, HO-1, and NQO1, resulted in significant improvements in renal function and increased survival
[120][131]. The naturally occurring antioxidant, nordihydroguaiaretic acid, prevents renal damage and apoptosis by inducing Nrf2 translocation in renal IRI
[122][133].