Diffuse large B-cell lymphoma (DLBCL) is the commonest form of lymphoid malignancy, with a prevalence of about 40% worldwide. The term DLBCL reflects the growth pattern and size of the neoplastic cells, which tend to diffusely efface the normal structure of the involved organ (most frequently the lymph node) and are provided with a diameter at least twice that of normal lymphocytes. Although during the last few years several distinct clinical-pathological categories of DLBCL have been reported in the literature, which are nowadays listed in the Revised 4th Edition of the WHO Classification of the Tumours of Haematopoietic and Lymphoid Tissues, about 80% of DLBCLs do not enter into any of these categories and are therefore termed not otherwise specified (NOS). DLBCL-NOS displays a quite variable morphology and only rarely consists of only one cytotype (centroblastic, immunoblastic or anaplastic). Thus, microscopic examination fails to define the cell of origin, prognostic indicators and novel potential therapeutic targets. The standard of care is the immuno-chemotherapy R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone), which cures up to 65% of patients. The remaining individuals with DLBCL-NOS experience resistant or relapsing disease and eventually die of it. This situation has promoted a huge number of studies focusing on the pathobiology of the tumour and based on high-throughput techniques, including gene expression profiling and next generation sequencing. In addition, attention has been focused on the mcroenvironmental composition, which can influence the behaviour and response to therapy of the tumour in conjunction with the molecular characteristics of neoplastic cells. The aim of the present review is to discuss the most recent acquisitions in the field of DLBCL-NOS based on the extensive application of molecular techniques, which paves the way to a more rational classification of the tumour along with the identification of effective prognostic indicators and novel therapeutic targets for ad hoc personalised approaches.
- Diffuse Large B-Cell Lymphomas
- Cell morphology
- Phenotype
- FISH analysis
- Gene expression profiling
- Next generation sequencing
- Cell of origin
- Micro-environment
- Gene mutation
1. Introduction
| Diffuse Large B-Cell Lymphoma (DLBCL): |
| DLBCL not otherwise specified (NOS) |
| Morphological variants |
| Centroblastic |
| Immunoblastic |
| Anaplastic |
| Other rare variants |
| Molecular subtypes |
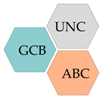
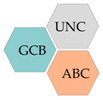
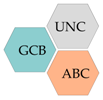
| Germinal centre B-cell subtype (GCB) |
| Activated B-cell subtype (ABC) |
| Other lymphomas of Large B-Cells: |
| T-cell/histiocyte-rich large B-cell lymphoma |
| Primary DLBCL of the CNS |
| Primary cutaneous DLBCL, leg type |
| EBV-positive DLBCL, NOS |
| EBV-positive mucocutaneous ulcer |
| DLBCL associated with chronic inflammation |
| Lymphomatoid granulomatosis |
| Large B-cell lymphoma with IRF4 rearrangement |
| Primary mediastinal (thymic) large B-cell lymphoma |
| Intravascular large B-cell lymphoma |
| ALK-positive large B-cell lymphoma |
| Plasmablastic lymphoma |
| HHV8-positive DLBCL |
| Primary effusion lymphoma |
| High-Grade B-Cell Lymphoma: |
| High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangement |
| High-grade B-cell lymphoma, not otherwise specified (NOS) |
2. Gene Expression Profiling
2.1. Cell of Origin (COO)
2.2. Key Genes
3. Tissue Microenvironment (TME)
4. Genetic Classification
4.1. Whole-Exome Sequencing (WES)-Based Studies
4.2. Targeted NGS and Bioinformatic-Based Studies
| Lacy et al. | Chapuy et al. | Schmitz et al. | Notes | ||||
|---|---|---|---|---|---|---|---|
| MYD88 | |||||||
| Genetics | GEP Signature | Related LNH | Targets | 5y-OS | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 |
C5 |  |
MCD | My-T-BCR dependent NF-kB; Immune evasion-MHC class I; Cell survival; BCL2 expression; Altered B-cell differentiation; G1-S cell cycle/p53 checkpoint; BCR: IgM >> IgG; IgVH 4-34++ |  |
B-cell activation NF-kB IRF4 MYC | MCD | Proliferation |
Primary extranodal DLBCL Transformed WM |  |
BCR-dep. NF-kB PI3K mTORC1 BCL2-BCLXL-MCL1 JAK1 MYD88 |
IRAK4 | Strongly associated with ABC. The most robust group in all reports. Contains the most primary PCNSL and testicular lymphoma. Poor prognosis. | |
| IRF4 | 40% All | 37% ABC | CD79B | |||||||||||
 |
N1 | NOTCH1 signaling Altered B cell differentiation BCR: IgM > IgG |
NOTCH Quiescence Plasma cells T cell-Myeloid-FDC |
NOTCH1 mutated CLL |
NOTCH1 Immune checkpoints |
27% All 22% ABC |
PIM1 | |||||||
 |
A53 | TP53 inactivation—DNA Damage Aneuploidy Immune evasion—B2M loss BCR: IgM >> IgG; IgVH 4-34++ |
p53 Immune low |
- | BCR-dep. NF-kB | 63% All 33% ABC 100% GCB |
ETV6 | |||||||
 |
BN2 | NOTCH2 signaling Altered B cell differentiation BCR-dependent NF-kB Immune evasion—CD70 loss Proliferation—Cyclin D3 BCR: IgM >> IgG; IgVH 4-34++ |
B-cell activation NF-kB NOTCH Proliferation |
MZL Transformed MZL |
BCR-dep. NF-kB PI3K mTORC1 BCL2 NOTCH2 |
67% All 76% ABC 100% GCB 38% UC |
CDKN2A | |||||||
 |
ST2 | JAK/STAT3 signaling NF-kB activation P2RY8-GNA13 activation Altered B cell differentiation BCR: IgM >> IgG |
GC B cell PI3K signaling JAK2 signaling Glycolysis Stromal |
NLPHL THRLBCL |
PI3K JAK2 |
84% All 81% GCB |
TBL1XR1 | |||||||
 |
MYC+ | Chromatin modification Anti-apoptosis PI3K signaling S1PR2-GNA13 inactivation Altered TFH interactions MYC (EZ-MYC+) BCR: IgG > IgM |
GC LZ (MYC−) GC IZ (MYC+) BCL6 (MYC+) TCF3 (both) TFH cells (MYC−) Stromal (MYC−) Immune low (MYC+) |
FL Transformed FL BL (EZB-MYC+) |
PI3K mTORC1 EZH2 BCL2-MCL1 |
48% MYC+ 82% MYC− |
BCL2 |  |
C3 |  |
EZB |  |
EZH2 | Strongly associated with GCB. Contains most transformed FLs and cases with a concurrent FL. Generally favorable prognosis, although enriched for cases of double-hit lymphoma and MHG. |
| BCL2 | ||||||||||||||
| BCL2 translocation | ||||||||||||||
| KMT2D | ||||||||||||||
| TNFRSF14 | ||||||||||||||
| MYC | CREBBP | |||||||||||||
| CREBBP2 | ||||||||||||||
| SOCS1/SGK1 |  |
C4 |  |
 |
CD83 | Predominantly GCB. Shares genetic and gene expression features of PMBCL. Associated with the most favorable prognosis. | ||||||||
| HIST1H1E | ||||||||||||||
| SGK1 | ||||||||||||||
| − | NFKBIA | |||||||||||||
| NFKBIE | ||||||||||||||
| SOCS1 | ||||||||||||||
| BRAF | ||||||||||||||
| TET2/SGK1 |  |
 |
 |
TET2 | A less strongly identifiable subtype. Has very strong similarity to SOCS1/SGK1 but differs by the addition of TET2 and BRAF and the lack of SOCS1 and CD83. Favorable prognosis. | |||||||||
| BRAF | ||||||||||||||
| SGK1 | ||||||||||||||
| KLHL6 | ||||||||||||||
| ID3 | ||||||||||||||
| NOTCH2 |  |
C1 |  |
BN2 |  |
BCL10 | Not associated with any COO. Shares mutational similarity to MZL but not enriched for transformed MZLs. Less strongly defined than other subgroups. | |||||||
| TNFAIP3 | ||||||||||||||
| NOTCH2 | ||||||||||||||
| BCL6 translocation | ||||||||||||||
| CCND3 | ||||||||||||||
| SPEN | ||||||||||||||
| UBE2A | ||||||||||||||
| CD70 | ||||||||||||||
| NEC |  |
 |
Other |  |
NOTCH1 | A default category, containing cases that could not be classified elsewhere and no detected mutation. Likely to also contain cases belonging to both NOTCH1 and TP53/CNA subgroups. | ||||||||
| REL amplification | ||||||||||||||
| TP53 | ||||||||||||||
 |
C2 |  |
 |
TP53 | Characterized by TP53 mutation and widespread copy number changes. | |||||||||
| Frequent deletions | ||||||||||||||
 |
C0 |  |
 |
No detected abnormalities | Cases with no detectable mutation were allocated to the NEC group. | |||||||||
 |
 |
N1 |  |
NOTCH1 | Characterized by NOTCH1 mutation, this was significantly elevated in Lacy’s NEC group but only mutated in 2.5% of samples. Associated with poor outcome. |
5. Open Issues and Perspectives
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; p. 2.
- Harris, N.L.; Jaffe, E.S.; Stein, H.; Banks, P.M.; Chan, J.K.; Cleary, M.L.; Delsol, G.; De Wolf-Peeters, C.; Falini, B.; Gatter, K.C.; et al. A revised European-American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 1994, 84, 1361–1392.
- Jaffe, E.S.; Harris, N.L.; Stein, H.; Vardiman, J. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2001.
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2008.
- Fisher, R.I.; Gaynor, E.R.; Dahlberg, S.; Oken, M.M.; Grogan, T.M.; Mize, E.M.; Glick, J.H.; Coltman, C.A.; Miller, T.P. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin’s lymphoma. N. Engl. J. Med. 1993, 328, 1002–1006.
- Harrysson, S.; Eloranta, S.; Ekberg, S.; Enblad, G.; Jerkeman, M.; Wahlin, B.E.; Andersson, P.-O.; Smedby, K.E. Incidence of relapsed/refractory diffuse large B-cell lymphoma (DLBCL) including CNS relapse in a population-based cohort of 4243 patients in Sweden. Blood Cancer J. 2021, 11, 9.
- Sehn, L.H.; Salles, G. Diffuse large B-cell lymphoma. N. Engl. J. Med. 2021, 384, 842–858.
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nat. Cell Biol. 2000, 403, 503–511.
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947.
- Nyman, H.; Jerkeman, M.; Karjalainen-Lindsberg, M.-L.; Banham, A.H.; Leppä, S. Prognostic impact of activated B-cell focused classification in diffuse large B-cell lymphoma patients treated with R-CHOP. Mod. Pathol. 2009, 22, 1094–1101.
- Wright, G.; Tan, B.; Rosenwald, A.; Hurt, E.H.; Wiestner, A.; Staudt, L.M. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9991–9996.
- Hans, C.P.; Weisenburger, D.D.; Greiner, T.C.; Gascoyne, R.D.; Delabie, J.; Ott, G.; Müller-Hermelink, H.K.; Campo, E.; Braziel, R.M.; Jaffe, E.S.; et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood 2004, 103, 275–282.
- Choi, W.W.L.; Weisenburger, D.D.; Greiner, T.C.; Piris, M.A.; Banham, A.H.; Delabie, J.; Braziel, R.M.; Geng, H.; Iqbal, J.; Lenz, G.; et al. A new immunostain algorithm classifies diffuse large B-cell lymphoma into molecular subtypes with high accuracy. Clin. Cancer Res. 2009, 15, 5494–5502.
- Colomo, L.; López-Guillermo, A.; Perales, M.; Rives, S.; Martínez, A.; Bosch, F.; Colomer, D.; Falini, B.; Montserrat, E.; Campo, E.; et al. Clinical impact of the differentiation profile assessed by immunophenotyping in patients with diffuse large B-cell lymphoma. Blood 2003, 101, 78–84.
- Muris, J.J.F.; Meijer, C.J.L.M.; Vos, W.; Van Krieken, J.H.J.M.; Jiwa, N.M.; Ossenkoppele, G.J.; Oudejans, J.J. Immunohistochemical profiling based on Bcl-2, CD10 and MUM1 expression improves risk stratification in patients with primary nodal diffuse large B cell lymphoma. J. Pathol. 2006, 208, 714–723.
- Meyer, P.N.; Fu, K.; Greiner, T.C.; Smith, L.M.; Delabie, J.; Gascoyne, R.D.; Ott, G.; Rosenwald, A.; Braziel, R.M.; Campo, E.; et al. Immunohistochemical methods for predicting cell of origin and survival in patients with diffuse large B-Cell lymphoma treated with rituximab. J. Clin. Oncol. 2011, 29, 200–207.
- Zinzani, P.L.; Dirnhofer, S.; Sabattini, E.; Alinari, L.; Piccaluga, P.P.; Stefoni, V.; Tani, M.; Musuraca, G.; Marchi, E.; Falini, B.; et al. Identification of outcome predictors in diffuse large B-cell lymphoma. Immunohistochemical profiling of homogeneously treated de novo tumors with nodal presentation on tissue micro-arrays. Haematologica 2005, 90, 341–347.
- Visco, C.; Li, Y.; Xu-Monette, Z.Y.; Miranda, R.N.; Green, T.M.; Li, Y.; Tzankov, A.; Wen, W.; Liu, W.-m.; Kahl, B.S.; et al. Comprehensive gene expression profiling and immunohistochemical studies support application of immunophenotypic algorithm for molecular subtype classification in diffuse large B-cell lymphoma: A report from the International DLBCL Rituximab-CHOP Consortium Program Study. Leukemia 2012, 26, 2103–2113.
- de Jong, D.; Rosenwald, A.; Chhanabhai, M.; Gaulard, P.; Klapper, W.; Lee, A.; Sander, B.; Thorns, C.; Campo, E.; Molina, T.; et al. Immunohistochemical prognostic markers in diffuse large B-cell lymphoma: Validation of tissue microarray as a prerequisite for broad clinical applications—A study from the Lunenburg Lymphoma Biomarker Consortium. J. Clin. Oncol. 2007, 25, 805–812.
- Gutiérrez-García, G.; Cardesa-Salzmann, T.; Climent, F.; González-Barca, E.; Mercadal, S.; Mate, J.L.; Sancho, J.M.; Arenillas, L.; Serrano, S.; Escoda, L.; et al. Gene-expression profiling and not immunophenotypic algorithms predicts prognosis in patients with diffuse large B-cell lymphoma treated with immunochemotherapy. Blood 2011, 117, 4836–4843.
- Scott, D.W.; Wright, G.W.; Williams, P.M.; Lih, C.-J.; Walsh, W.; Jaffe, E.S.; Rosenwald, A.; Campo, E.; Chan, W.C.; Connors, J.M.; et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014, 123, 1214–1217.
- Scott, D.W.; Mottok, A.; Ennishi, D.; Wright, G.W.; Farinha, P.; Ben-Neriah, S.; Kridel, R.; Barry, G.S.; Hother, C.; Abrisqueta, P.; et al. Prognostic significance of diffuse large B-cell lymphoma cell of origin determined by digital gene expression in formalin-fixed paraffin-embedded tissue biopsies. J. Clin. Oncol. 2015, 33, 2848–2856.
- Jais, J.P.; Molina, T.J.; Ruminy, P.; Gentien, D.; Reyes, C.; Scott, D.W.; Rimsza, L.M.; Wright, G.; Gascoyne, R.D.; Staudt, L.M.; et al. Reliable subtype classification of diffuse large B-cell lymphoma samples from GELA LNH2003 trials using the Lymph2Cx gene expression assay. Haematologica 2017, 102, e404–e406.
- Gifford, G.; Gabrielli, S.; Gill, A.; Greenwood, M.; Wong, K.; Best, G.; Nevell, D.; Mcllroy, K.; Kliman, D.; Ilmay-Gillespie, L.; et al. Lymphoma cell-of-origin assignment by gene expression profiling is clinically meaningful across broad laboratory contexts. Br. J. Haematol. 2018, 181, 272–275.
- Derenzini, E.; Mazzara, S.; Melle, F.; Motta, G.; Fabbri, M.; Bruna, R.; Agostinelli, C.; Cesano, A.; Corsini, C.A.; Chen, N.; et al. A 3-gene signature based on MYC, BCL-2 and NFKBIA improves risk stratification in diffuse large B-cell lymphoma. Haematologica 2020. Online ahead of Print.
- Staiger, A.M.; Ziepert, M.; Horn, H.; Scott, D.W.; Barth, T.F.E.; Bernd, H.-W.; Feller, A.C.; Klapper, W.; Szczepanowski, M.; Hummel, M.; et al. Clinical impact of the cell-of-origin classification and the MYC/BCL2 dual expresser status in diffuse large B-cell lymphoma treated within prospective clinical trials of the german high-grade non-Hodgkin’s lymphoma study group. J. Clin. Oncol. 2017, 35, 2515–2526.
- Vitolo, U.; Trněný, M.; Belada, D.; Burke, J.M.; Carella, A.M.; Chua, N.; Abrisqueta, P.; Demeter, J.; Flinn, I.; Hong, X.; et al. Obinutuzumab or rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone in previously untreated diffuse large B-cell lymphoma. J. Clin. Oncol. 2017, 35, 3529–3537.
- Vitolo, U.; Witzig, T.E.; Gascoyne, R.D.; Scott, D.W.; Zhang, Q.; Jurczak, W.; Özcan, M.; Hong, X.; Zhu, J.; Jin, J.; et al. ROBUST: First report of phase III randomized study of lenalidomide/R-CHOP (R2-CHOP) vs placebo/R-CHOP in previously untreated ABC-type diffuse large B-cell lymphoma. Hematol. Oncol. 2019, 37, 36–37.
- Green, T.M.; Young, K.H.; Visco, C.; Xu-Monette, Z.Y.; Orazi, A.; Go, R.S.; Nielsen, O.; Gadeberg, O.V.; Mourits-Andersen, T.; Frederiksen, M.; et al. Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J. Clin. Oncol. 2012, 30, 3460–3467.
- Hu, S.; Xu-Monette, Z.Y.; Tzankov, A.; Green, T.; Wu, L.; Balasubramanyam, A.; Liu, W.; Visco, C.; Li, Y.; Miranda, R.N.; et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: A report from The International DLBCL Rituximab-CHOP Consortium Program. Blood 2013, 121, 4021–4031.
- Ziepert, M.; Lazzi, S.; Santi, R.; Vergoni, F.; Granai, M.; Mancini, V.; Staiger, A.; Horn, H.; Löffler, M.; Pöschel, V.; et al. A 70% cut-off for MYC protein expression in diffuse large B cell lymphoma identifies a high-risk group of patients. Haematologica 2020, 105, 2667–2670.
- Chan, W.C. Using gene expression profiling to move beyond MYC/BCL2 rearrangements in high-grade lymphoma. J. Clin. Oncol. 2019, 37, 175–177.
- Sha, C.; Barrans, S.; Cucco, F.; Bentley, M.A.; Care, M.A.; Cummin, T.; Kennedy, H.; Thompson, J.S.; Uddin, R.; Worrillow, L.; et al. Molecular high-grade B-cell lymphoma: Defining a poor-risk group that requires different approaches to therapy. J. Clin. Oncol. 2019, 37, 202–212.
- Ennishi, D.; Jiang, A.; Boyle, M.; Collinge, B.; Grande, B.M.; Ben-Neriah, S.; Rushton, C.; Tang, J.; Thomas, N.; Slack, G.W.; et al. Double-hit gene expression signature defines a distinct sub-group of germinal center B-cell-like diffuse large B-cell lymphoma. J. Clin. Oncol. 2019, 37, 190–201.
- Jost, P.J.; Ruland, J. Aberrant NF-kappaB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707.
- Davies, A.; Cummin, T.E.; Barrans, S.; Maishman, T.; Mamot, C.; Novak, U.; Caddy, J.; Stanton, L.; Kazmi-Stokes, S.; McMillan, A.; et al. Gene-expression profiling of bortezomib added to standard chemoimmunotherapy for diffuse large B-cell lymphoma (REMoDL-B): An open label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 649–662.
- Bu, R.; Hussain, A.R.; Al-Obaisi, K.A.S.; Ahmed, M.; Uddin, S.; Al-Kuraya, K.S. Bortezomib inhibits proteasomal degradation of IκBα and induces mitochondrial dependent apoptosis in activated B-cell diffuse large B-cell lymphoma. Leuk. Lymphoma 2014, 55, 415–424.
- Mottok, A.; Wright, G.; Rosenwald, A.; Ott, G.; Ramsower, C.; Campo, E.; Braziel, R.M.; Delabie, J.; Weisenburger, D.D.; Song, J.Y.; et al. Molecular classification of primary mediastinal large B-cell lymphoma using routinely available tissue specimens. Blood 2018, 132, 2401–2405.
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323.
- Alizadeh, A.A.; Gentles, A.J.; Alencar, A.J.; Liu, C.L.; Kohrt, H.E.; Houot, R.; Goldstein, M.J.; Zhao, S.; Natkunam, Y.; Advani, R.H.; et al. Prediction of survival in diffuse large B-cell lymphoma based on the expression of 2 genes reflecting tumor and microenvironment. Blood 2011, 118, 1350–1358.
- Meyer, P.N.; Fu, K.; Greiner, T.; Smith, L.; Delabie, J.; Gascoyne, R.; Ott, G.; Rosenwald, A.; Braziel, R.; Campo, E.; et al. The stromal cell marker SPARC predicts for survival in patients with diffuse large B-cell lymphoma treated with rituximab. Am. J. Clin. Pathol. 2011, 135, 54–61.
- Keane, C.; Gill, D.; Vari, F.; Cross, D.; Griffiths, L.; Gandhi, M. CD4+ tumor infiltrating lymphocytes are prognostic and independent of RIPI in patients with DLBCL receiving R-CHOP chemo-immunotherapy. Am. J. Hematol. 2013, 88, 273–276.
- Abdou, A.G.; Asaad, N.; Kandil, M.; Shabaan, M.; Shams, A. Significance of stromal-1 and stromal-2 signatures and biologic prognostic model in diffuse large B-cell lymphoma. Cancer Biol. Med. 2017, 14, 151–161.
- Ciavarella, S.; Vegliante, M.C.; Fabbri, M.; De Summa, S.; Melle, F.; Motta, G.; De Iuliis, V.; Opinto, G.; Enjuanes, A.; Rega, S.; et al. Dissection of DLBCL microenvironment provides a gene expression-based predictor of survival applicable to formalin-fixed paraffin-embedded tissue. Ann. Oncol. 2018, 29, 2363–2370.
- Staiger, A.M.; Altenbuchinger, M.; Ziepert, M.; Kohler, C.; Horn, H.; Huttner, M.; Hüttl, K.S.; Glehr, G.; Klapper, W.; Szczepanoswki, M.; et al. Emed Demonstrator Project; German High Grade Non-Hodgkin’s Lymphoma Study Group (DSHNHL): A novel lymphoma-associated macrophage interaction signature (LAMIS) provides robust risk prognostication in diffuse large B-cell lymphoma clinical trial cohorts of the DSHNHL. Leukemia 2020, 34, 543–552.
- Tripodo, C.; Zanardi, F.; Iannelli, F.; Mazzara, S.; Vegliante, M.; Morello, G.; Di Napoli, A.; Mangogna, A.; Facchetti, F.; Sangaletti, S.; et al. A spatially resolved dark-versus light-zone microenvironment signature subdivides germinal center-related aggressive B cell lymphomas. iScience 2020, 23, 101562.
- Ennishi, D.; Takata, K.; Béguelin, W.; Duns, G.; Mottok, A.; Farinha, P.; Bashashati, A.; Saberi, S.; Boyle, M.; Meissner, B.; et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov. 2019, 9, 546–563.
- Kotlov, N.; Bagaev, A.; Revuelta, M.V.; Phillip, J.M.; Cacciapuoti, M.T.; Antysheva, Z.; Svekolkin, V.; Tikhonova, E.; Miheecheva, N.; Kuzkina, N.; et al. Clinical and biological subtypes of B-cell lymphoma revealed by microenvironmental signatures. Cancer Discov 2021. Online ahead of Print.
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.L.; et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell 2017, 171, 481–494.e15.
- Bolen, C.R.; Klanova, M.; Trneny, M.; Sehn, L.H.; He, J.; Tong, J.; Paulson, J.N.; Kim, E.; Vitolo, U.; Di Rocco, A.; et al. Prognostic impact of somatic mutations in diffuse large B-cell lymphoma and relationship to cell-of-origin: Data from the phase III GOYA study. Haematologica 2019, 105, 2298–2307.
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690.
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407.
- Xu-Monette, Z.Y.; Wu, L.; Visco, C.; Tai, Y.C.; Tzankov, A.; Liu, W.-M.; Montes-Moreno, S.; Dybkaer, K.; Chiu, A.; Orazi, A.; et al. Mutational profile and prognostic significance of TP53 in diffuse large B-cell lymphoma patients treated with R-CHOP: Report from an International DLBCL Rituximab-CHOP Consortium Program Study. Blood 2012, 120, 3986–3996.
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.L.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771.
- Ennishi, D.; Healy, S.; Bashashati, A.; Saberi, S.; Hother, C.; Mottok, A.; Chan, F.C.; Chong, L.; Abraham, L.; Kridel, R.; et al. TMEM30A loss-of-function mutations drive lymphomagenesis and confer therapeutically exploitable vulnerability in B-cell lymphoma. Nat. Med. 2020, 26, 577–588.
- Wright, G.W.; Huang, D.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell 2020, 37, 551–568.e14.
- Morin, R.D.; Scott, D.W. DLBCL subclassification: Divide and conquer? Blood 2020, 135, 1722–1724.
- Tabanelli, V.; Melle, F.; Motta, G.; Mazzara, S.; Fabbri, M.; Corsini, C.; Gerbino, E.; Calleri, A.; Sapienza, M.R.; Abbene, I.; et al. Evolutionary crossroads: Morphological heterogeneity reflects divergent intra-clonal evolution in a case of high-grade B-cell lymphoma. Haematologica 2020, 105, e432–e436.
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.M.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct biological subtypes and patterns of genome evolution in lymphoma revealed by circulating tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155.
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse large B-cell lymphoma genotyping on the liquid biopsy. Blood 2017, 129, 1947–1957.
- Kurtz, D.M.; Esfahani, M.S.; Scherer, F.; Soo, J.; Jin, M.C.; Liu, C.L.; Newman, A.M.; Dührsen, U.; Hüttmann, A.; Casasnovas, O.; et al. Dynamic risk profiling using serial tumor biomarkers for personalized outcome prediction. Cell 2019, 178, 699–713.e19.