Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Xueyuan Sun and Version 3 by Vivi Li.
In this entry, we discuss the use of the alkylating agent temozolomide (TMZ) in the treatment of IDH-mutant gliomas. We describe the challenges associated with TMZ in clinical (drug resistance and tumor recurrence) and preclinical settings (variabilities associated with in vitro models) in treating IDH-mutant glioma.
- IDH-mutant glioma
- TMZ
- combination therapy
1. Introduction
Gliomas are the most common primary malignant tumors in the central nervous system. Grade 2 and 3 gliomas are referred to as lower grade gliomas (LGG) and harbor mutations in the isocitrate dehydrogenase (IDH) gene [1]. IDH-mutant gliomas have a slower growth rate and longer survival than IDH wild type (IDH-wt) tumors [1][2][1,2]. IDH-mutant gliomas are classified into two subgroups based on the presence (astrocytoma) or absence (oligodendroglioma) of chromosome arms 1p/19q [3] and histological criteria [4]. Recently, the European Association of Neuro-Oncology (EANO) stratified IDH-mutant gliomas into three WHO grades: oligodendroglioma, WHO grade 2 or 3; astrocytoma, WHO grade 2 or 3; astrocytoma, WHO grade 4 [5]. Although slower growing (at a rate of ~4–5 mm per year [6]), the majority of IDH-mutant LGGs eventually undergo malignant progression due to activation of the PI3K/mTOR pathway as a result of PTEN loss [7][8][7,8] or enhanced PDGF signaling [9]. Detailed molecular diagnostic markers, and other common molecular and pathway alterations in IDH-mutant gliomas are summarized in Table 1.
Table 1. Molecular diagnostic markers and common genetic alterations in IDH-mutant glioma.
| Category | Alterations | Oligodendroglioma WHO Grade 2 | Oligodendroglioma WHO Grade 3 | Astrocytoma WHO Grade 2/3 | Astrocytoma WHO Grade 4 | |
|---|---|---|---|---|---|---|
| Diagnostic markers | IDH1 or IDH2 mutation | Present | Present | Present | Present | |
| G-CIMP | Present | Present | Present | Present | ||
| ATRX | Inactivated | Inactivated | ||||
| 1p (FUBP1) / 19q (CIC) codeletion | Present | Present | ||||
| TERT | Activated | Activated | ||||
| 9p21 (CDKN2A/B) | Inactivated | |||||
| Necrosis and/or microvascular proliferation | Present | |||||
| Other genomic alterations | TP53 | Inactivated | Inactivated | |||
| Myc | Activated | |||||
| TCF12 | Inactivated | |||||
| 10q (PTEN/MGMT) | Inactivated | |||||
| Signaling pathways | Activation of PI3K signaling through loss of PTEN and gain of mTOR | |||||
| Activation of cell cycle signaling through gain of CDK4, CDK6 and cyclin E2 | ||||||
ATRX, Alpha-thalassemia/mental retardation, X-linked; CDKN2A/B, Cyclin-dependent kinase inhibitor 2A/B; CDK, Cyclin-dependent kinases; CIC, Capicua transcriptional repressor; FUBP1, Far upstream element binding protein 1; G-CIMP, cytosine-phosphate-guanine (CpG) island methylator phenotype; IDH, Isocitrate dehydrogenase; MGMT, Methylguanine-DNA-Methyltransferase; PI3K, Phosphoinositide 3-kinase; PTEN, Phosphatase and tensin homolog; TCF12, Transcription factor 12; TERT, Telomerase reverse transcriptase.
The IDH gene encodes the enzyme isocitrate dehydrogenase, which converts isocitrate to α-ketoglutarate (α-KG). α-KG an intermediate of the tricarboxylic acid (TCA) cycle that contributes to the production of NADPH. NADPH is necessary to reduce oxidized glutathione to glutathione, which directly neutralizes free radicals and reactive oxygen species (ROS). Overall, 65% of total NADPH in glioblastoma (GBM) is driven by the enzymatic activity of IDH, which is reduced to 38% when IDH is mutated [10]. There are three IDH isoforms, IDH1, IDH2, and IDH3, which are encoded by different genes. IDH1 is localized in the cytosol and peroxisomes, while IDH2 and IDH3 are located in the mitochondria. Among them, IDH1 is most frequently mutated in gliomas and harbors a monoallelic missense mutation of arginine to histidine at position 132 (IDH1R132H) at the catalytic site of the enzyme. IDH mutation produces a neomorphic enzyme that converts α-KG to D-(R)-2-hydroxyglutarate (2-HG), leading to the accumulation of 2-HG in the tumor [11]. The oncometabolite 2-HG is a competitive inhibitor of α-KG-dependent enzymes, including DNA demethylases (family of TET enzymes) and histone demethylases (family of Jumonji enzymes) [12][13][12,13]. This inhibition modifies the epigenetic status of histones and DNA, resulting in a plethora of cellular changes, including DNA hypermethylation [14] and altered histone methylation [15] (Figure 1).
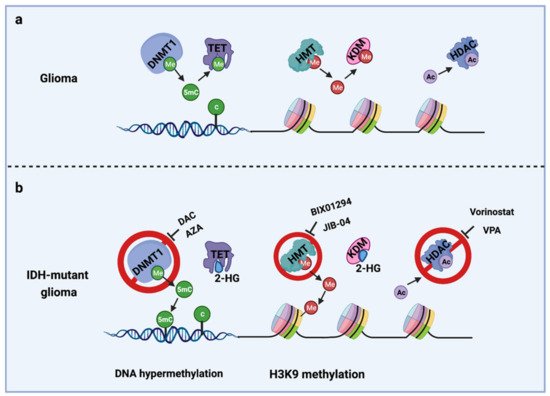
Figure 1. Epigenetic alterations induced by IDH mutations and potential drug targets for TMZ combination therapy. (a) Cellular epigenetic regulation without IDH mutation; (b) cellular epigenetic alterations in IDH-mutant gliomas. AZA, 5-azacitidine; DAC, Decitabine; DNMT1, DNA methyltransferase 1; HDAC, Histone deacetylase; 2-HG, D-(R)-2-hydroxyglutarate; HMT, Histone methyltransferase; KDM, Histone demethylase; TET, Ten-eleven translocation methylcytosine dioxygenases; VPA, Valproic acid; 5mC, 5-Mehylcytosine. Me: Methyl group; Ac, Acetyl group.
Treatment of LGGs includes surgery, radiation, and chemotherapy with either procarbazine/lomustine/vincristine (PCV) or temozolomide (TMZ). Here, we focus on the use of TMZ in IDH-mutant LGGs. First, we will present the effect of IDH mutation on cellular metabolism, epigenetic modifications, and the targeted therapies associated with these alterations. Second, we will discuss the use of TMZ in the treatment of IDH-mutant gliomas, including its toxicity, TMZ-associated molecular signature in tumor recurrence, and drug resistance, and discuss the synthetic lethality opportunities that emerge with TMZ treatment of IDH-mutant gliomas. Third, we will discuss the challenges of using TMZ to treat IDH-mutant gliomas in the preclinical setting, including non-consensus TMZ dosage and regimen, variable methods in measuring cell viability, and difficulties in culturing IDH-mutant glioma cell lines.
2. Temozolomide Treatment in IDH-Mutant Gliomas
Standard-of-care treatment for gliomas includes maximal surgical resection, possibly followed by radiotherapy (RT) and chemotherapy with PCV or TMZ. Several randomized clinical trials [16][39] investigating dosing (EORTC 22844 [17][40]) and timing (EORTC 22845 [18][41]) for RT in LGGs show that RT alone provides no significant benefit for overall survival. Similarly, TMZ alone showed no significant difference in progression-free survival in patients with LGGs compared with the efficacy of RT (EORTC 22033-26033 [19][42]). However, RT combined with TMZ or PCV resulted in an overall survival benefit in patients [20][43]. IDH-mutant oligodendrogliomas benefit from the addition of PCV to RT (RTOG 9802 [21][22][44,45], RTOG 9402 [23][46], EORTC 26951 [24][47], and NOA-04 [25][48]), while RT plus TMZ treatment shows more benefit in astrocytomas in clinical (EORTC 26053-22054 (CATNON) [26][49], RTOG 0424 [27][50]), and retrospective [28][51] studies. The interim analysis of the CATNON trial indicate a trend toward benefit with concurrent TMZ in IDH-mutant tumors, but not in IDH-wt gliomas. Thus, EANO recommends RT + TMZ for the treatment of newly diagnosed astrocytomas. For oligodendrogliomas, EANO recommends RT + PCV for initial treatment, and TMZ is only recommended for recurrent tumors not being pre-treated with TMZ [5]. Currently, there are no mature data comparing TMZ and PCV or their combination with radiation for LGGs. The ongoing clinical trial ALLIANCE-N0577-CODEL comparing RT + TMZ with RT + PCV for anaplastic oligodendrogliomas with 1p/19q co-deletion could potentially provide a more definitive comparison between the two regimens [29][52]. Both PCV and TMZ have been associated with grade 3 and 4 hematologic toxicities. Clinicians largely suggest TMZ to patients instead of PCV (>85%) [30][53], considering the relative difficulty of administering intravenous vincristine and the greater toxicity of PCV [31][54], whereas TMZ is easy to administer and generally well tolerated [20][32][43,55]. The ongoing phase III EORTC-1635-BTG (Wait or Treat?) is a randomized phase III trial comparing early adjuvant treatment with radiotherapy and adjuvant temozolomide to active surveillance in patients with resected IDH-mutant astrocytoma. Here, we summarize the current challenges related to TMZ in gliomas with a particular focus on IDH-mutant tumors.2.1. Mechanisms of TMZ Toxicity
TMZ is administered orally in capsules at a dose of 150–200 mg/m2 for 5 out of 28 days for 6–12 cycles [5]. TMZ is a lipophilic DNA alkylating prodrug, and the cytotoxicity of TMZ is mediated by the addition of methyl groups to DNA. TMZ is an imidazotetrazine derivative of dacarbazine. Under neutral pH and aqueous conditions, it spontaneously decarboxylates to generate 5-(3-methyltriazen-1-yl)-imidazole-4-carboxamide (MTIC), which is further degraded to 4-amino-5-imidazole-carboxamide (AIC), and a highly reactive methyldiazonium ion that acts as a DNA methylating species [33][56] (Figure 23a). About 60–80% of methyl groups are added at DNA guanine residues (N7-MeG), 10–20% of the methyl groups are added at adenine (N3-MeA), and 10% of methyl groups at guanine (O6-MeG) [34][57] (Figure 23b). Single damaged bases, N7-MeG and N3-MeA, are readily removed by the rapid and efficient base excision repair (BER) system before replication. Therefore, the key toxic insult of TMZ is attributed to the O6-meG lesions [35][36][58,59].
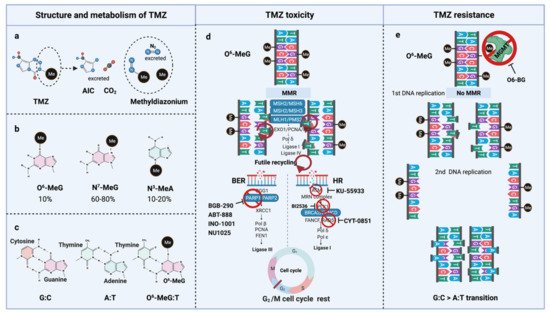
Figure 23. TMZ molecular structure, metabolism, toxicity, and resistance. (a) TMZ structure and metabolism, (b) DNA methylation upon TMZ, (c) DNA base mispair upon DNA methylation, (d) mechanism of TMZ toxicity with intact MMR, BER, and HR, (e) mechanism of TMZ resistance with functional MGMT and non-functional MMR. AIC, 4-Amino-5-imidazolecarboxamide; BER, Base-excision repair; HR, Homologous repair; MGMT, O6-methylguanine-DNA-methyltransferase; MMR, Mismatch repair; MLH, MutL homologue; MSH, MutS homologue; PMS, Post-meiotic segregation; TMZ, Temozolomide.
O6-meG is considered the most genotoxic base modification due to the subsequent nucleotide mispairing with thymine (T) instead of cytosine (C) during DNA replication (Figure 23c). During replication, DNA polymerase inserts T opposite O6-meG. The mismatch repair (MMR) system can detect and repair these mismatches through the MutS and MutL complexes. The MutS recognition complex, including MutSα (an MSH2/MSH6 heterodimer) and MutSb (MSH2/ MSH3 heterodimer), identifies base–base mismatches and binds the O6-meG: T mismatch. Upon binding to the mismatch, the MutS complex recruits MutL (MLH1/PMS2 dimer) to the site of DNA damage. Together, these proteins excise a stretch of single-stranded DNA (ssDNA) containing the mispaired T, creating a gap in the DNA, while leaving the O6-meG adduct on the template strand intact [37][60]. DNA polymerase fills the gap by reinserting T opposite O6-meG, triggering another round of MMR which leads to repeated attempts to repair the same base T. This futile MMR cycling and accumulation of ssDNA gaps lead to successively longer DNA reinsertion and excision, which generates double strand breaks (DSBs) in subsequent rounds of replication and induce cell cycle arrest in G2/M phase, apoptosis and autophagy [38][61]. Thus, it needs two cell divisions for the emergence of TMZ cytotoxicity [39][62] (Figure 23d).
However, O6-meG lesions can be directly removed by O6-methylguanine DNA methyltransferase (MGMT) through covalent transfer, a process that effectively repairs the alteration prior to replication (Figure 23e). MGMT promoter methylation is a predictive biomarker of TMZ response in GBM. In general, the repair of O6-meG depends on the number of MGMT molecules per cell and the rate of MGMT regeneration [40][63]. In summary, the cytotoxicity from TMZ depends on low MGMT levels [41][64] and an intact MMR pathway [42][65].
2.2. Maintenance of TMZ Sensitivity
Efforts have focused on maintaining TMZ sensitivity by reducing MGMT levels or attenuating the activity of the BER and HR pathways for the duration of TMZ treatment to prevent resistance.
MGMT. O6–benzylguanine (O6–BG) is a potent inhibitor of the repair protein O6–alkylguanine–DNA alkyltransferase (AGT) that effectively inhibits MGMT activity by suicide inactivation. O6-BG binds and inactivates AGT, and until new AGT protein is synthesized, the cells have increased sensitivity to TMZ [43][44][45][66,67,68], leading to several clinical trials combining O6-BG and TMZ [44][46][47][48][49][50][67,69,70,71,72,73]. A phase II study showed that one-day dosing of O6-BG plus TMZ restored TMZ sensitivity in patients with TMZ-resistant anaplastic IDH-mutant gliomas [50][73] (Table 2).
Table 2. Preclinical and clinical studies of combination therapy of TMZ with DNA damage repair pathway inhibitors.
| Targeting DNA Damage Repair | Synergistic with TMZ | Preclinical Model | Clinical Trial | Arms | Tumor Type | Phase | Year | |
|---|---|---|---|---|---|---|---|---|
| MGMTi | O6BG + TMZ | GBM PDX [45]; astrocytoma or GBM patient [44] | GBM PDX [68]; astrocytoma or GBM patient [67] | NCT00006474 | O6-BG + TMZ | Astrocytoma | I | 2001–2004 |
| NCT00389090 | O6-BG + TMZ | Gliomas | II | (2006–2009)Terminated | ||||
| NCT00613093 | O6-BG + TMZ | GBM | II | 2002–2008 | ||||
| NCT00275002 | O6-BG + TMZ | Pediatrichigh-grade gliomas | II | 2006–2010 | ||||
| PARPi | Olaparib + TMZ | U87-IDH mutant, U251-IDH mutant cell lines [51] | U87-IDH mutant, U251-IDH mutant cell lines [81] | NCT03212742 | Olaparib + TMZ + IMRT | GBM | I/IIa | 2017–2022 |
| NCT04394858 | Olaparib + TMZ | Pheochromocytoma and paraganglioma | II | 2020–2023 | ||||
| Veliparib (ABT-888)+TMZ | GBM BTICs and xenografts [52] | GBM BTICs and xenografts [80] | NCT01026493 | ABT-888 + TMZ | Recurrent GBM | I/II | 2010–2016 | |
| NCT01514201 | RT+ ABT-888 + TMZ | Children with newly diagnosed DIPG | I/II | 2012–2018 | ||||
| NCT02152982 | veliparib + TMZ vs. placebo + TMZ | GBM | II/III | 2014–2020 | ||||
| NCT03581292 | RT + TMZ + veliparib | GBM | II | 2018–2024 | ||||
| Pamiparib (BGB-290) + TMZ | GBM, GL261 murine glioma cells xenografts [53] | GBM, GL261 murine glioma cells xenografts [82] | NCT03150862 | BGB-290 + RT vs. BGB-290 + TMZ | GBM | 1b/2 | 2017–2021 | |
| NCT03914742 | BGB-290 + TMZ | Recurrent IDH mutant glioma | I/II | 2020–2023 | ||||
| NCT03914742 | BGB-290 + TMZ | IDH mutant glioma | I | 2019–2027 |
BTICs, Brain tumor initiating cells; DIPG, Diffuse intrinsic pontine gliomas; GBM, Glioblastoma multiforme; MGMTi, Methylguanine-DNA-Methyltransferase inhibitor; PARPi, Poly (ADP-ribose) polymerase inhibitor; PDX, patient-derived xenograft; TMZ, Temozolomide.
MMR. MMR is regulated by multiple signaling pathways and responds to many stimuli [54][74]. Mutations in MMR genes and loss of MMR function are frequently detected in tumor samples [55][75]. Nevertheless, there is little research on how to maintain MMR integrity or directly increase MMR activity. One study showed that EGFRvIII expression and MAPK activation lead to increased MMR and therefore TMZ sensitivity [56][76], with direct clinical relevance that anti-EGFRvIII and anti-MAPK strategies should be used with caution in combination with TMZ.
BER. Many efforts have been made to target the downstream ssDNA repair and HR system to maintain TMZ sensitivity. One target from the base-excision repair (BER) pathway is poly (ADP-ribose) polymerase 1 (PARP1), which facilitates DNA repair by binding to single-strand breaks and recruiting DNA repair proteins to the site of damage [57][77]. PARP inhibitors (PARPi), INO-1001 [58][78], NU1025 [59][79], ABT-888 (veliparib) [52][51][80,81] and pamiparib [53][82], restored sensitivity in TMZ resistant glioma cells and xenografts. Several PARPi in combination with TMZ in GBM have been registered for clinical trials [60][61][62][83,84,85]. However, there are only two phase I clinical trials testing the efficacy of pamiparib (BGB-290) in combination with TMZ in newly diagnosed or recurrent IDH1/2-mutant gliomas (NCT03914742 and NCT03749187), and its clinical efficacy in IDH-mutant gliomas has yet to be demonstrated [63][86] (Table 2).
HR. One target from the HR pathway is the homologous recombinase RAD51. RAD51 is involved in DNA strand exchange between homologous DNA sequences. Exogenously expressed mutant IDH1 increases RAD51-driven HR and leads to increased TMZ resistance, and RAD51 knockdown increases the sensitivity of glioma cells to TMZ [64][87]. Several drug screens have identified inhibitors of RAD51 [65][88], but there is only one active clinical trial directly targeting RAD51 using a small molecule inhibitor CYT-0851 (NCT03997968). Another HR target is PLK1 (Polo-like kinase 1), which phosphorylates BRCA1 [66][89] and RAD51 [67][90] to promote homologous recombination. The combination of TMZ with a PLK1 inhibitor, BI2536, significantly suppressed the growth of IDH1-mutant glioma tumors [68][91], induced G2/M arrest, and suppressed cell proliferation and sphere formation [69][92]. Due to the toxicity of the small molecule inhibitor, knockdown of PLK1 using a small interfering RNA (siRNA) was combined with TMZ for glioma treatment, which showed enhanced anti-tumor activity both in vitro and in vivo [70][93]. However, limited success has been reported in preclinical studies with PLK1 inhibitors. ATM (ataxia telangiectasia mutated), a protein kinase that is a central mediator of responses to DNA double-strand breaks in cells [71][94], can also be targeted therapeutically. KU-55933, an ATM inhibitor, enhances the cytotoxic effects of TMZ in IDH1-mutant glioma cell lines [72][95].
Tumor Microtubes. Recently, there has been increasing evidence that tumor microtubes (TM) are an important mechanism of therapy resistance in gliomas. Gliomas interconnect and communicate through a network of TMs [73][96]. TM-connected glioma cells can self-repair, and are resistant to radiotherapy [73][96] and TMZ [74][97]. Inhibition of gap junctions with INI-0602 sensitizes primary GBM cells to TMZ [75][98], which shows the importance of pharmacological inhibition of the TM network. Furthermore, disrupting TM-based networks with meclofenamate (MFA) [76][99] sensitized primary glioblastoma cells to TMZ. The fact that TMs are more abundant in astrocytomas than in oligodendrogliomas [73][96], might explain why astrocytoma patients have a better response to TMZ than oligodendroglioma patients. A phase I/II trial evaluating safety as well as feasibility of a combined MFA-TMZ approach in relapsed MGMT-methylated glioblastoma (“MecMeth” EudraCT2021-000708-39) is being initiated in Germany [76][99].
2.3. TMZ-Associated Hypermutation
The first report investigating the effect of TMZ in the treatment of LGGs [77][100] showed that although most tumors exhibited initial chemosensitivity, the majority of tumors resumed progressive growth within a year of TMZ treatment, with astrocytomas (20/33) exhibiting a higher regrowth rate than oligodendrogliomas (5/30), implying that astrocytomas acquire accelerated TMZ resistance than oligodendrogliomas. Another long-term follow-up study showed that most oligodendrogliomas resumed growth within 3 years after TMZ [78][101].
TMZ resistance can be acquired either by elevated MGMT levels [79][80][102,103] or by mutations in the MMR machinery [81][82][83][84][85][104,105,106,107,108] that prevent futile MMR cycles at unrepaired O6-meG lesions. In the absence of MGMT-mediated repair in conjunction with deficient MMR, long-term TMZ treatment causes cells to accumulate G:C>A:T transitions throughout the genome, resulting in a hypermutator phenotype in recurrent tumors [40][86][63,109] (Figure 23e). Long-term TMZ treatment could also inactivate MMR pathway genes leading to hypermutation [86][109].
TMZ-induced hypermutation is observed more frequently in IDH-mutant than in IDH-wt gliomas [87][88][110,111]. However, it is not clear which subtype is more prone to develop the hypermutator phenotype. Reports from paired primary and TMZ-treated recurrent tumors show that astrocytomas have a higher rate of hypermutation [88][89][111,112], while data from random patient samples show that TMZ-induced hypermutation is more prevalent in oligodendrogliomas [86][87][109,110]. TMZ-induced hypermutation has been associated with a worse prognosis [89][112]; however, a larger cohort from the Glioma Longitudinal Analysis (GLASS) consortium shows no differences in overall survival between hypermutators and non-hypermutators [88][111].
Increased tumor mutation burden correlates with an elevated neoantigen load, indicating the potential to induce a durable response to immunotherapy [90][113]. However, current data show no discernible differences in the extent of immunoediting between initial and TMZ-treated relapsed hyper-mutated gliomas [88][111], and neoantigens from the recurrent hypermutators have relatively poor immunogenic qualities which may result in a weak anti-tumor T-cell response and likely a poor response to immunotherapy [86][109]. A current clinical trial is evaluating the immune-activating antibody pembrolizumab (MK-3475) in recurrent malignant gliomas that exhibit the hypermutator phenotype (NCT02658279).
2.4. TMZ-Induced Cellular Adaptations and Combination Therapy in IDH-Mutant Glioma
In addition to the known MGMT activity and DNA repair pathways in conferring TMZ resistance, efforts have been made to understand genetic, epigenetic, or metabolic adaptations following TMZ treatment. This knowledge could lead to synthetic lethal targeted strategies, with combinations of targeted therapies to circumvent some resistance mechanisms to delay or prevent malignant progression and recurrence [91][92][114,115]. Previous research has mainly focused on TMZ resistance in IDH-wt GBM [93][94][95][116,117,118], and here we summarize the current literatures on TMZ resistance mechanisms and therapeutic options in IDH-mutant gliomas.
2.4.1. Genetic Mutations Associated with TMZ Treatment in IDH Mutant Glioma
Direct comparison of the genomic landscape of gliomas at initial diagnosis and recurrence has provided insight into the genomic alterations that may be associated with tumor recurrence after TMZ. Analysis of copy number alterations (CNAs) from primary IDH-mutant and IDH-wt gliomas of all grades revealed amplification of cyclins and cyclin-dependent kinase genes in IDH-mutant gliomas [96][119] (Table 1). A cohort of six pairs of initial untreated and TMZ treated recurrent IDH-mutant gliomas showed that recurrent tumors have driver mutations that activate retinoblastoma (Rb) and mammalian target of rapamycin (mTOR) pathways [89][112], which might drive malignant progression. The mTOR inhibitors such as rapamycin (RAPA) [97][120] have been reported to enhance TMZ-induced autophagic death of GBM cells and inhibition of the Akt-mTOR signaling pathway with amlexanox enhances TMZ-induced anti-tumor effects in preclinical GBM models [98][121]. An orally bioavailable dual PI3K/mTOR inhibitor, XL765 (voxtalisib), produced additive toxicity when combined with TMZ in genetically diverse GBM xenografts [99][122]. A phase I clinical trial (NCT00704080) demonstrated a favorable safety profile and a moderate inhibition of the PI3K/mTOR pathway in all glioma subtypes [100][123]. Sequential treatment of TMZ followed by PX-866, a PI3K inhibitor, inhibited TMZ-induced autophagy survival and enhanced apoptosis in GBM cells [101][124]. These findings suggest that PI3K/mTOR/Rb signaling pathways can be targeted separately or together to prevent tumor progression after TMZ treatment.
However, a study by the GLASS consortium comparing 23 pairs of untreated primary and TMZ-treated recurrent IDH-mutant gliomas [88][111] did not identify specific driver mutations associated with TMZ resistance. Across all cohorts, the hotspot IDH1R132H mutation was not lost during progression and remained clonal in all progressed tumors [87][110], providing a good rationale for IDHR132H vaccines for targeted therapies.
CRISPR-based screening enables sensitive detection of drug-gene interactions directly in human cells. Although no genome-wide CRISPR-Cas9 screen has been performed in IDH-mutant glioma models, results from GBM patient-derived lines [102][125] and GBM adherent lines [103][126] indicated that mismatch repair (MMR) and HR pathways are involved in TMZ resistance. In addition to the MMR pathways, an interesting molecular alteration detected in the human GBM cell line is NRF2 activation, and inhibition of NRF2 enhanced the anti-tumor effect of TMZ in glioma cells [104][127]. Since NRF2 is important for maintaining the redox balance in IDH-mutant gliomas and increasing ROS has been shown to augment chemosensitivity in IDH-mutant glioma [105][106][128,129], it is plausible that NRF2 inhibitors in combination with TMZ may be promising for the treatment of IDH-mutant gliomas.
Pathway analysis from RNA-seq data obtained from preclinical GBM models showed that epithelial–mesenchymal transition, Wnt signaling, and immune response were the most significantly activated pathways in TMZ-resistant cell lines [107][130]. In addition, negative regulation of telomere maintenance via telomerase was enriched in TMZ-sensitive glioma cell lines. A synergistic effect of a combination treatment of TMZ and a telomerase inhibitor, BIBR1532, was observed in in vitro models of GBM [107][130]. Whether telomerase inhibitors in combination with TMZ have an anti-tumor effect in IDH-mutant gliomas requires further investigation.
2.4.2. Epigenetic Alterations upon TMZ Treatment
DNA methylation. Preclinical studies have shown that high TMZ concentration leads to a short-term increase in total 5-methylcytosine (hypermethylation), while repeated low TMZ doses lead to DNA hypomethylation [108][131]. This indirect effect on DNA methylation status may partly explain why 5-azacytidine (AZA) in combination with TMZ has a better anti-tumor effect in IDH1-mutant glioma patient-derived xenograft (PDX) models [109][132]. A phase I clinical trial of AZA combined with TMZ in patients with unresectable or metastatic soft tissue sarcoma or malignant mesothelioma shows that both drugs can be administered at their full dose without dose-limiting toxicities (NCT00629343) [110][133]. DAC, another DNA methyltransferase inhibitor, has been shown to potentiate TMZ treatment by enhancing the effects of DNA damage [111][134] and DNA mismatch repair [112][135] in GBM. A phase I/II clinical trial of the combination of DAC and TMZ in metastatic melanoma has shown that DAC can be safely added to extended-schedule TMZ and leads to improved response rates and progression-free survival (PFS) and overall survival (OS) rates in patients [113][136]. Due to the dose-dependent effect of TMZ on epigenetic modifications, further studies are needed to determine appropriate treatment regimens for the combination of TMZ and epigenetic therapy to achieve optimal clinical benefit.
Histone methylation. An inhibitor of histone methyltransferase (HMT) G9a, BIX01294, also exerted a synergistic effect with TMZ in GBM [114][137], possibly by enhancing the autophagy pathway. JIB-04, a novel inhibitor of Jumonji demethylases [115][138], synergized strongly with TMZ [116][117][139,140] in GBM in vitro and in vivo. Since IDH-mutant gliomas exhibit increased histone H3 lysine 9 (H3K9) methylation [118][141], the combination of G9a and TMZ may be a potential therapeutic target in IDH-mutant gliomas.
Histone acetylation. Several histone deacetylase (HDAC) inhibitors synergize with TMZ. For example, vorinostat [119][142] is well tolerated in combination with TMZ in GBM patients in a phase II trial (NCT00731731) [120][143] and are currently evaluated in combination with RT. Valproic acid (VPA) [121][144], another HDAC inhibitor with concurrent RT and TMZ are also well tolerated in GBM in a phase II study (NCT00302159) [122][123][145,146]. In a phase I clinical trial, a triple agent of dual epigenetic therapy, a combination of DAC, panobinostat (an HDAC inhibitor) and TMZ was well-tolerated, and its further efficacy is currently being evaluated in a phase II trial (NCT00925132) [124][147]. As epigenetic alterations may represent a global mechanism of resistance in cancer [125][148], preclinical experiments and clinical trials will clarify whether epigenetic therapy can act synergistically with TMZ in IDH-mutant gliomas.
2.4.3. Metabolic Changes after TMZ Treatment
Glutamate. Previous studies have indicated that long-term TMZ treatment leads to changes in amino acid metabolism in preclinical models of oligodendroglioma [126][149]. Other reports identified that increased glutamate/glutamine/GLX (the sum of glutamate and glutamine) levels could be an early indication of response to TMZ treatment in IDH1-mutant gliomas [127][128][150,151]. IDH-mutant tumors have lower glutamate levels; thus, combination therapy of GLS inhibitor and TMZ may provide a greater benefit in IDH-mutant gliomas. Loss of xCT/SLC7A11, the glutamate exchanger that plays a role in ferroptosis, leads to increased vulnerability to TMZ [129][130][152,153]. These studies suggest that the effect of TMZ can be potentiated by ferroptosis inducing agents such as erastin and sorafenib. Another study showed that the addition of the glutaminase inhibitor CB-839 to TMZ significantly reduced aspartate and glutamate levels in an IDH-mutant patient-derived glioma xenograft model [131][154]. A phase I clinical study is currently evaluating the combination of CB-839, RT, and TMZ in IDH-mutated diffuse or anaplastic astrocytomas (NCT03528642) [132][155].
Phospholipid. The late-stage autophagy inhibitors chloroquine (CQ) and bafilomycin A1 (BAF) restore phospholipid levels and inhibit clonogenicity of IDH-mutant glioma cells. CQ enhances the cytotoxic effects of TMZ in GBM [133][156], and its clinical impact is being investigated in a phase I trial (NCT02378532). It is possible that the combination of CQ and TMZ disrupts the phospholipid balance and has greater synergistic effect in IDH-mutant gliomas.
NAD+. TMZ treatment leads to NAD+ consumption driven by PARP activation, as NAD+ is a known PARP cofactor. In IDH1-mutant cells with already low basal NAD+ levels, this surge in consumption leads to a further reduction in NAD+. Importantly, this metabolic imbalance introduces a window of hypervulnerability to NAD+ biosynthesis inhibitors [134][30]. Indeed, combined TMZ and NAMPT inhibition showed better efficacy in vivo than either agent alone [135][157]. Although the role of PARP in TMZ resistance is paradoxical as PARP needs to be inhibited to suppress its DNA repair function to maintain TMZ sensitivity but should be activated to drive NAD+ scarcity for its anti-tumor effect in IDH-mutant cells. The ongoing clinical trials of PARPi + TMZ in IDH-mutant glioma (NCT03914742, NCT03749187, NCT04394858, NCT01026493) [136][158] will give us a clear answer in the near future (Table 3). NAD+ is used for making poly (ADP-ribose) (PARylation) to recruit DNA repair factors [137][159]. PARylation is eventually degraded by PAR glycohydrolase (PARG), and NAD+ is regenerated. Therefore, combining TMZ with a PARG inhibitor COH34 leads to a scarcity of available NAD+, which is highly effective against IDH-mutant gliomas [138][160]. We expect PARG inhibitors with better toxicity profiles to be developed for preclinical and clinical trials in the near future.
Glucose. TMZ treatment has been shown to increase the expression of glucose transporters (GLUTs) [139][140][161,162], which triggers higher glycolytic activity and decreases the response to TMZ treatment, while inhibition of GLUT/SLC2A enhances the effect of TMZ [140][162]. Combination treatment with TMZ and paclitaxel (Taxol), a microtubule inhibitor, sensitized Taxol-resistant glioma cells via inhibition of glucose metabolism [141][163] and is currently in a phase II trial for the treatment of patients with metastatic melanoma (NCT01009515) [142][164]. Metformin, another metabolic inhibitor, alters both whole-body and cellular energy metabolism, and also shows a synergistic effect when combined with TMZ in GBM [143][165]. Trehalose, a natural disaccharide of glucose, combined with TMZ reduced clonogenicity and enhanced autophagic effects in melanoma cells [144][166]. Whether targeting glucose metabolism enhances the efficacy of TMZ in IDH-mutant gliomas requires further investigation.
A triple therapy combination with TMZ, CQ, and rapamycin decreased mitochondrial function and induced lysosome-dependent apoptotic cell death [145][167], suggesting that combinatorial targeting of metabolic and genetic alterations may be a good therapeutic option in cancer therapy in the future. Here, we summarize in Table 34 current vulnerable targets that could potentially be combined with TMZ in preclinical and clinical settings. Some of these have been tested only in GBM or other tumor entities but have the potential to be applicable to IDH-mutant gliomas as well.
Table 34. Preclinical and clinical studies of TMZ combined with therapies targeting cancer metabolism
| Metabolic Target | Combination Therapy | Preclinical Model | Clinical Trial | Arms | Tumor Type | Phase | Year | |
|---|---|---|---|---|---|---|---|---|
| NAD+ | NAMPT inhibitor + TMZ | IDH1 mutant glioma lines [134] | IDH1 mutant glioma lines [30] | NCT00724841 | GEM1777 + TMZ | Metastatic melanoma | I/II | 2008–2010 (Terminated) |
| Glutamine | CB-839 + TMZ | GBM164 (IDH mutant) and GBM6 (IDH wt) PDX [131] | GBM164 (IDH mutant) and GBM6 (IDH wt) PDX [154] | NCT03528642 | CB-839 + RT + TMZ | Astrocytoma | 1b | 2018–2022 |
| Oxidative phosphorylation | MET + TMZ | GBM PDX [143] | GBM PDX [165] | NCT01430351 | MET + TMZ vs. mefloquine + TMZ vs. memantine + TMZ | GBM | I | 2011–2022 |
| Phospholipid | CQ + TMZ | GBM cell lines [133] | GBM cell lines [156] | NCT02378532 | CQ + RT + TMZ | GBM | I | 2016–219 |
| Multiple metabolites | Paclitaxel + TMZ | GBM cell lines [141] | GBM cell lines [163] | NCT01009515 | Carboplatin + Paclitaxel + TMZ | Metastatic melanoma | II | 2009–2015 (Terminated) |
| MET + CQ | NCT02496741 | MET + CQ | IDH mutant glioma | 1b | 2015–2019 |
CQ, Chloroquine; MET, Metformin; NAMPT, Nicotinamide phosphoribosyltransferase; PDX, Patient-derived xenografts; RT, Radiotherapy; TMZ, Temozolomide.