Neoplastic cells typically activate one of the two Telomere Maintenance Mechanisms
(TMM) to maintain their telomeres during uncontrolled proliferation. Most tumors reactivate
telomerase, a high-fidelity DNA transferase with reverse transcriptase activity. The prevalence of telomerase positive cancers is at 80–90% of all malignancies. A significant percentage of neoplasias activate the second type of TMM, called alternative
lengthening of telomeres (ALT), to achieve replicative immortality and telomere
elongation. ALT is commonly thought to occur in about 10–20% of all tumors. Unlike the TEL+ tumors, which rely on the enzymatic activity of a single enzyme,
ALT relies on many DNA damage response (DDR) proteins, including those involved in
the homology-dependent repair (HDR) pathway.
- alternative lengthening of telomeres
- cancers
- ALT biomarkers
- ALT prevalence
- ATRX
- DAXX
1. Commonly Used ALT Biomarkers
[1]
[3]
[6]
[7]
Table 1.
| Biomarkers | Telo-FISH Nuclear Foci | APBs | TIFs | Telomerase Activity | C-circles | tSCE | Telomere Heterogeneity |
|---|
| ALT+ | √ | √ | √ | X | √ | √ | √ |
| TEL+ | X | X | X | √ | X | X | X |
| Ever-Shorter Telomeres | X | X | X | X | X | X | X |
ALT+: alternative lengthening of telomere (ALT) positive. TEL+: telomerase (TEL) positive. X: negative or relatively low; √: positive or relatively high; No shading: tissue sections; Light shading: tissue homogenate; Dark shading: tissue culture. Telo-FISH: Telomere (Telo) fluorescent in-situ hybridization (FISH). APBs: ALT-associated acute promyelocytic leukemia bodies. TIFs: telomere dysfunction-induced foci. tSCE: telomere sister chromatid exchange.
[8]
[9]
[8]
[9]
[9]
[14]
[9]
[16]
[17]
[9]
[20]
[24]
[25]
[26]
[26]
[27]
[29]
[27]
[30]
2. The prevalence of ALT in a variety of human cancers.
Table 2.
| Tumor | % ALT+ | Range * | Total Tumors Tested, n |
|---|
| Bone | |||
| Chondrosarcoma | 48% | N/A | 31 |
| Ewing Sarcoma | 0% | N/A | 62 |
| Osteosarcoma | 63% | 49–86% | 287 |
| Breast | |||
| 36–77% | |||
| 174 | |||
*: Minimum and maximum of horizontal range is per the mean ALT+ prevalence acquired of CCA, APB, Telo-FISH (alone), and TRF, respectively. N/A: not applicable – horizontal range could not be generated as this tumor was only assessed via one biomarker.
3. The prognosis of ALT+ vs. TEL+ cancers.
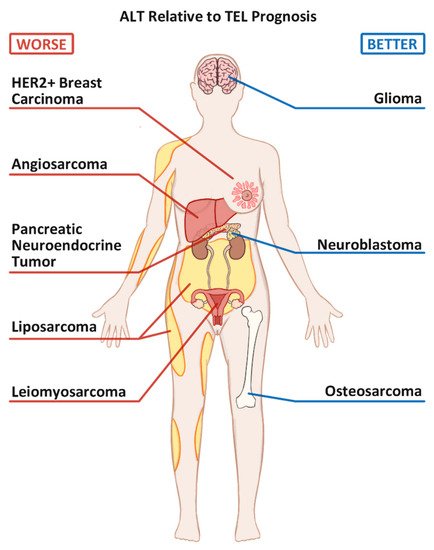
Figure 1.
4. Mutation Status of ATRX and DAXX in ALT cancers
4. Mutation Status of ATRX and DAXX
ATRX
DAXX
ATRX
DAXX
[40]
Table 3.
| Tumor | ATRX Loss Only, n | DAXX Loss Only, n | Both ATRX and DAXX Loss, n | Total Tumors Tested, n | Total Mutant % * | ATRX/DAXX Mutant Cases that are ALT+, % | ALT+ Cases that are ATRX Mutant, % |
|---|
| Bone | |||||||||||
| Chondrosarcoma | 0 ** | N/A *** | N/A | 15 | 0% | N/A | N/A | ||||
| Ewing Sarcoma | 0 | N/A | N/A | 12 | 0% | N/A | N/A | ||||
| Osteosarcoma | 17 | 0 | 0 | 71 | 24% | 100% | 58% | ||||
| Breast | |||||||||||
| Overall | 4% | ||||||||||
| Breast Carcinoma | 0 | 2–8% | 0552 | ||||||||
| 0 | 96 | 0% | N/A | N/A | HER2+ Breast Carcinoma | 18% | 14–21% | 50 | |||
| CNS | Central Nervous System | ||||||||||
| Glioma | 403 | ||||||||||
| 7 | N/A | 1607 | 26% | 74% | Overall | 20% | 10–26% | 4386 | |||
| 71% | |||||||||||
| Neuroendocrine | Glioma | 30% | 13–69% | 912 | |||||||
| Neuroblastoma (NB) | 83 | 1 | 0 | 1052 | 8% | 92% | 67% | NF1 loss-associated glioma | 44% | 29–69% | 167 |
| High Risk NB | 25 | 0 | 0 | 165 | 15% | N/A | 100% | Astrocytoma (Overall) | 23% | 10–78% | |
| PanNET | 218 | 153 | 2231 | ||||||||
| 23 | 1223 | 32% | 96% | 86% | Diffuse astrocytoma (grade II) | 55% | 27–100% | 95 | |||
| Soft Tissue | Anaplastic astrocytoma (grade III) | 65% | 21–92% | ||||||||
| Angiosarcoma | 16 | 118 | |||||||||
| 0 | 2 | 77 | 21% | N/A | 88% | Adult glioblastoma (grade IV) | 16% | 11–25% | 862 | ||
| Leiomyoma | 6 | 1 | 0 | 206 | 3% | 43% | 67% | Pediatric glioblastoma (grade IV) | 39% | 12–56% | 89 |
| Leiomyosarcoma | 103 | 4 | 0 | 311 | 34% | 83% | 56% | Oligodendroglioma | 17% | ||
| Liposarcoma (LPS) | 39 | 0–25% | 178 | ||||||||
| N/A | 203 | 20% | 100% | 78% | Oligoastrocytoma | 60% | 11–72% | 48 | |||
| Well differentiated LPS | 0 | N/A | N/A | 6 | 0% | N/A | N/A | Ependymoma | 0% | ||
| Dedifferentiated LPS | N/A | 28 | 260 | ||||||||
| 1 | 0 | 52 | 56% | 100% | 93% | Medulloblastoma | 7% | 2–22% | 237 | ||
| Myxoid LPS | 0 | N/A | N/A | 55 | 0% | N/A | N/A | Choroid plexus carcinomas | 23% | N/A | 31 |
| Pleomorphic LPS | 11 | N/A | N/A | 27 | 41% | 100% | 63% | Neuroendocrine | |||
| Undifferentiated Pleomorphic Sarcoma | |||||||||||
| 32 | N/A | N/A | 87 | 37% | 96% | 55% | Neuroblastoma | 24% | 18–47% | 843 | |
| Pancreatic Neuroendocrine Tumor (PanNET) | 32% | 21–61% | 1152 | ||||||||
| Soft Tissue | |||||||||||
| Angiosarcoma | 23% | 11–24% | 79 | ||||||||
| Leiomyoma | 3% | 0–3% | 217 | ||||||||
| Leiomyosarcoma | 60% | 52–78% | 331 | ||||||||
| Liposarcoma | 27% | 25–29% | 566 | ||||||||
| Undifferentiated Pleomorphic Sarcoma | 59% |
*: Total Mutant % = (ATRX loss + DAXX loss + Both loss)/Total Tumors; **: 0 indicates that at least one study assessed for the gene, but gene loss was not observed in any samples; ***: N/A indicates that the data was not available or the study did not assess for the gene of interest. ATRX: α-thalassemia/mental retardation syndrome X-linked (ATRX) gene. DAXX: death-domain associated protein gene.
References
- Bryan, T.M.; Englezou, A.; Gupta, J.; Bacchetti, S.; Reddel, R.R. Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J. 1995, 14, 4240–4248.
- Murnane, J.; Sabatier, L.; Marder, B.; Morgan, W. Telomere dynamics in an immortal human cell line. EMBO J. 1994, 13, 4953–4962.
- Londoño-Vallejo, J.A.; Der-Sarkissian, H.; Cazes, L.; Bacchetti, S.; Reddel, R.R. Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res. 2004, 64, 2324–2327.
- Tokutake, Y.; Matsumoto, T.; Watanabe, T.; Maeda, S.; Tahara, H.; Sakamoto, S.; Niida, H.; Sugimoto, M.; Ide, T.; Furuichi, Y. Extra-Chromosomal Telomere Repeat DNA in Telomerase-Negative Immortalized Cell Lines. Biochem. Biophys. Res. Commun. 1998, 247, 765–772.
- Cesare, A.J.; Kaul, Z.; Cohen, S.B.; E Napier, C.; Pickett, H.A.; Neumann, A.A.; Reddel, R.R. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat. Struct. Mol. Biol. 2009, 16, 1244–1251.
- Yeager, T.R.; Neumann, A.A.; Englezou, A.; Huschtscha, L.I.; Noble, J.R.; Reddel, R.R. Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res. 1999, 59, 4175–4179.
- Takai, H.; Smogorzewska, A.; de Lange, T. DNA Damage Foci at Dysfunctional Telomeres. Curr. Biol. 2003, 13, 1549–1556.
- Claude, E.; Decottignies, A. Telomere maintenance mechanisms in cancer: Telomerase, ALT or lack thereof. Curr. Opin. Genet. Dev. 2020, 60, 1–8.
- Henson, J.D.; Reddel, R.R. Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett. 2010, 584, 3800–3811.
- Pan, X.; Drosopoulos, W.C.; Sethi, L.; Madireddy, A.; Schildkraut, C.L.; Zhang, D. FANCM, BRCA1, and BLM cooperatively resolve the replication stress at the ALT telomeres. Proc. Natl. Acad. Sci. 2017, 114, E5940–E5949.
- Lu, R.; O’Rourke, J.J.; Sobinoff, A.P.; Allen, J.A.M.; Nelson, C.B.; Tomlinson, C.G.; Lee, M.; Reddel, R.R.; Deans, A.J.; Pickett, H.A. The FANCM-BLM-TOP3A-RMI complex suppresses alternative lengthening of telomeres (ALT). Nat. Commun. 2019, 10, 2252.
- Pan, X.; Chen, Y.; Biju, B.; Ahmed, N.; Kong, J.; Goldenberg, M.; Huang, J.; Mohan, N.; Klosek, S.; Parsa, K.; et al. FANCM suppresses DNA replication stress at ALT telomeres by disrupting TERRA R-loops. Sci. Rep. 2019, 9, 19110.
- Feng, E.; Batenburg, N.L.; Walker, J.R.; Ho, A.; Mitchell, T.R.H.; Qin, J.; Zhu, X.-D. CSB cooperates with SMARCAL1 to maintain telomere stability in ALT cells. J. Cell Sci. 2020, 133, 133.
- Sobinoff, A.P.; Pickett, H.A. Alternative Lengthening of Telomeres: DNA Repair Pathways Converge. Trends Genet. 2017, 33, 921–932.
- Draskovic, I.; Arnoult, N.; Steiner, V.; Bacchetti, S.; Lomonte, P.; Londoño-Vallejo, A. Probing PML body function in ALT cells reveals spatiotemporal requirements for telomere recombination. Proc. Natl. Acad. Sci. USA 2009, 106, 15726–15731.
- Loe, T.K.; Li, J.S.Z.; Zhang, Y.; Azeroglu, B.; Boddy, M.N.; Denchi, E.L. Telomere length heterogeneity in ALT cells is maintained by PML-dependent localization of the BTR complex to telomeres. Genes Dev. 2020, 34, 650–662.
- Dellaire, G. The Nuclear Protein Database (NPD): Sub-nuclear localisation and functional annotation of the nuclear proteome. Nucleic Acids Res. 2003, 31, 328–330.
- Min, J.; Wright, W.E.; Shay, J.W. Clustered telomeres in phase-separated nuclear condensates engage mitotic DNA synthesis through BLM and RAD52. Genes Dev. 2019, 33, 814–827.
- Zhang, H.; Zhao, R.; Tones, J.; Liu, M.; Dilley, R.L.; Chenoweth, D.M.; Greenberg, R.A.; Lampson, M.A. Nuclear body phase separation drives telomere clustering in ALT cancer cells. Mol. Biol. Cell 2020, 31, 2048–2056.
- Pickett, H.A.; Cesare, A.J.; Johnston, R.L.; Neumann, A.A.; Reddel, R.R. Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J. 2009, 28, 799–809.
- Cerone, M.A.; Autexier, C.; Londoño-Vallejo, J.A.; Bacchetti, S. A human cell line that maintains telomeres in the absence of telomerase and of key markers of ALT. Oncogene 2005, 24, 7893–7901.
- Fasching, C.L.; Bower, K.; Reddel, R.R. Telomerase-Independent Telomere Length Maintenance in the Absence of Alternative Lengthening of Telomeres–Associated Promyelocytic Leukemia Bodies. Cancer Res. 2005, 65, 2722–2729.
- Marciniak, R.A.; Cavazos, D.; Montellano, R.; Chen, Q.; Guarente, L.; Johnson, F.B. A Novel Telomere Structure in a Human Alternative Lengthening of Telomeres Cell Line. Cancer Res. 2005, 65, 2730–2737.
- Henson, J.D.; A Hannay, J.; McCarthy, S.W.; A Royds, J.; Yeager, T.R.; A Robinson, R.; Wharton, S.B.; A Jellinek, D.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225.
- Zhang, J.-M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cell Rep. 2019, 26, 955–968.e3.
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.-M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Netto, G.J.; Epstein, J.I.; Lotan, T.L.; Westra, W.H.; et al. Prevalence of the Alternative Lengthening of Telomeres Telomere Maintenance Mechanism in Human Cancer Subtypes. Am. J. Pathol. 2011, 179, 1608–1615.
- Henson, J.D.; Cao, Y.; I Huschtscha, L.; Chang, A.C.; Au, A.Y.M.; Pickett, H.A.; Reddel, R.R. DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat. Biotechnol. 2009, 27, 1181–1185.
- Zhang, T.; Zhang, Z.; Shengzhao, G.; Li, X.; Liu, H.; Zhao, Y. Strand break-induced replication fork collapse leads to C-circles, C-overhangs and telomeric recombination. PLoS Genet. 2019, 15, e1007925.
- Mazzucco, G.; Huda, A.; Galli, M.; Piccini, D.; Giannattasio, M.; Pessina, F.; Doksani, Y. Telomere damage induces internal loops that generate telomeric circles. Nat. Commun. 2020, 11, 1–11.
- Henson, J.D.; Lau, L.M.; Koch, S.; La Rotta, N.M.; Dagg, R.A.; Reddel, R.R. The C-Circle Assay for alternative-lengthening-of-telomeres activity. Methods 2017, 114, 74–84.
- Mason-Osann, E.; Gali, H.; Flynn, R.L. Resolving Roadblocks to Telomere Replication. Methods Mol. Biol. 2019, 1999, 31–57.
- Kim, J.; Sun, C.; Tran, A.D.; Chin, P.-J.; Ruiz, P.D.; Wang, K.; Gibbons, R.J.; Gamble, M.J.; Liu, Y.; Oberdoerffer, P. The macroH2A1.2 histone variant links ATRX loss to alternative telomere lengthening. Nat. Struct. Mol. Biol. 2019, 26, 213–219.
- Clynes, D.; Jelinska, C.; Xella, B.; Ayyub, H.; Scott, C.; Mitson, M.; Taylor, S.S.; Higgs, D.R.; Gibbons, R.J. Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat. Commun. 2015, 6, 7538.
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277.
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558.
- Shuai, J.; Shi, G.; Zhang, L.; Yuanling, J.; Jiang, Y.; Jiang, S.; Ma, W.; Zhao, Y.; Songyang, Z.; Huang, J. Switch telomerase to ALT mechanism by inducing telomeric DNA damages and dysfunction of ATRX and DAXX. Sci. Rep. 2016, 6, srep32280.
- Heaphy, C.M.; De Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science 2011, 333, 425.
- Jiao, Y.; Shi, C.; Edil, B.H.; De Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199–1203.
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.W.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Khuong-Quang, D.-A.; Tönjes, M.; et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231.
- Lovejoy, C.A.; Li, W.; Reisenweber, S.; Thongthip, S.; Bruno, J.; De Lange, T.; De, S.; Petrini, J.H.J.; Sung, P.A.; Jasin, M.; et al. Loss of ATRX, Genome Instability, and an Altered DNA Damage Response Are Hallmarks of the Alternative Lengthening of Telomeres Pathway. PLoS Genet. 2012, 8, e1002772.