The gut microbiota plays an important role in maintaining homeostasis in the human body, and the disruption of these communities can lead to compromised host health and the onset of disease.
- microbiota
- probiotics
- lactic acid bacteria
- cancer
- colorectal cancer
1. Introduction
Currently, colorectal cancer (CRC) is the third most common cancer worldwide, with more than one million new cases and 600,000 deaths each year [1]. There are two types of CRC: colitis-associated, caused by the presence of a mutation in the TP53 gene, and sporadic, caused by a mutation in the adenomatous polyposis coli (APC) gene. However, genetic factors play a relatively minor role in cancer development (<10% to 30%); instead, cancer risk is greatly influenced by extrinsic (e.g., environmental) factors such as infectious agents, antibiotic administration, high-fat diets, red meat consumption, and a deficiency in fiber intake [2][3][2,3]. All of these components are known to alter gut microbiota and induce dysbiosis [4], defined as perturbations in commensal communities that can lead to the deficient education of the host immune system and the subsequent development of immune-mediated diseases. Dysbiosis can be categorized into three types: (i) loss of beneficial species, (ii) expansion of pathobionts or potentially harmful species, and (iii) loss of overall microbial diversity [5]. All three types of dysbiosis have been observed in CRC patients.
One of the means by which healthy gut microbiota may exert their anticancer effects is through the beneficial metabolites they produce, which can have antioxidant and anti-inflammatory properties, regulate bowel barrier function, act as vitamins, and represent a source of energy. Instead, the gut microbiota of CRC patients can have direct pro-tumorigenic effects; for example, a gavage of fecal samples from CRC patients was observed to promote intestinal carcinogenesis in both germ-free and conventional mice [6]. Recent research into probiotics, and into microbiota more generally, has yielded promising outcomes and has demonstrated the serious potential of these assemblages as co-adjuvants in colon cancer therapies.
2. Gut Microbiota
The microbiota, also referred to as the microflora, is defined as the entire population of microbes present within the human body, which principally includes bacteria, archaea, and eukarya [7], as well as viruses [8]. The quantity of these microorganisms is staggering. The human gastrointestinal tract alone can host nearly 100 trillion (1014) microorganisms [9], a number nearly three times greater than the total number of cells in the entire human body (recently recalculated as 3.7 × 1013) [10]. From a physiological point of view, the microbiota makes up about 2% of an adult’s body mass, almost equivalent to the size of the human brain or liver [11], which has led some researchers to refer to the microbiota as the forgotten human organ [12][13][12,13]. Many essential body processes require the presence of these diverse and numerous microorganisms, as they provide the host with nutrients, metabolize indigestible compounds, and can help in the defense against colonization by opportunistic pathogens, as well as possess immune-modulatory properties [14].
With the use of next-generation DNA sequencing technologies and metagenomic analysis, it has been shown that the gut microbiota of vertebrates is composed of approximately 500–1000 different bacterial species, of which 98% are represented by two dominant phyla, Bacteroidetes and Firmicutes [15][16][17][15,16,17]. One of the most surprising discoveries has been the fact that the number of genes in the gut microbiota is approximately 100 times larger than the human genome and appears to represent a co-evolutionary relationship [17]. Due to the numerous interactions among different microbial species, human host cells, and the external environment, the microbiota can also be conceptualized as being a dynamic ecological community [18]. In this sense, a dynamic equilibrium of the microbiota in the human body is necessary for health, which can be disrupted by environmental factors and external stimuli such as the use of antibiotics, illness, stress, aging, bad dietary habits, and lifestyle [19]. These alterations frequently result in microbial imbalances—dysbiosis—with direct links to multiple pathological conditions [20].
For example, between 2018 and 2021, a search for “microbiota dysbiosis” and “diseases” in PubMed returned 5,617 published articles describing links between dysbiosis of the gut microbiota and diseases such as obesity [21], autism spectrum disorders [22], cardiovascular diseases [23], diarrhea [24], alcoholic liver disease [25], acute-on-chronic liver failure [26], arthritis [27], lung diseases [28], autoimmune diseases [29], lupus erythematosus [30], coeliac disease [31], intestinal inflammatory diseases such as Crohn’s disease, colitis and irritable bowel syndrome (IBD) [32], and colorectal cancer [33] (these account for 531 citations by themselves).
3. CRC and Gut Microbiota
Compared to healthy individuals, CRC patients harbor a distinct mucosa-associated microbiota. For instance, the effect of CRC on the microbiota is generally characterized by an increase in microbial diversity that seems to progress with cancer development—late CRC samples (stage III and stage IV) generally display higher richness levels than early CRC samples do (stage I and stage II) [34]. At the phylum level, increasing numbers of Bacteroidetes, Firmicutes, and Fusobacteria and decreasing numbers of Proteobacteria in mucosa-associated microbiota are observed. At the genus level, CRC progression tends to be associated with the proliferation of Fusobacterium, Peptostreptococcus, Streptococcus, and Ruminococcus and a decline in Lactobacillus and Granulicatella [34][35][34,35] (Figure 1). Differences in mucosa-associated microbiota can also be observed before the appearance of a cancerous state. For example, Flemer et al. [36] detected significant differences between the mucosa-associated microbiota from subjects with polyps and from healthy controls, suggesting that the gut microbiota is involved in cancer development from a very early stage. Another large-cohort multi-omics dataset indicated that shifts in the microbiome and metabolome occur from the very early stages of the development of colorectal cancer, which could be of possible etiological and diagnostic importance [37]. Accordingly, it has been reported that a long-term exposure (≥2 months) to antibiotics in early to middle adulthood is associated with an increased risk for colorectal adenoma at the age of 60 [38]. Interestingly, the microbiota alterations observed in CRC patients are not restricted to the tumor site; they can also be seen in the surrounding healthy tissue. Indeed, in multiple cohorts, the mucosa-associated microbiota from paired samples of tumor tissue and nearby nontumor mucosa was similar with regard to both individual taxa and the overall microbiota composition [34][35][39][40][34,35,39,40].
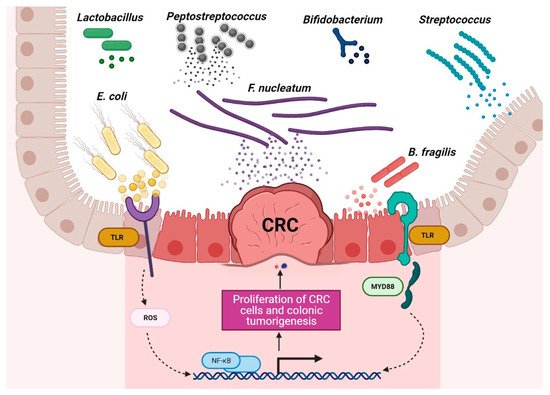
Figure 1. Schematic of the roles of probiotic and harmful bacteria in CRC context. Disruption of the gut microbiota balance is associated with CRC development, and regulation of probiotic bacteria is associated with CRC remission. (The figure was created with Biorender.com).
A comparative study of coupled fecal and mucosal samples demonstrated that, although fecal microbiota only partially reflects the community at the mucus layer, differences due to CRC are still evident in fecal samples [36]. Thus, this noninvasive approach is the most commonly used sampling method in gut microbiome studies. Fecal samples from CRC patients differ significantly from those of healthy subjects, both in microbial richness and community composition. In fecal samples, CRC development is usually associated with an increase in pro-inflammatory or pathogenic species belonging to phyla Proteobacteria and Fusobacteria and a decrease in beneficial species of phylum Firmicutes [41]. As observed with the mucosa-associated microbiota, the fecal microbiota of CRC patients is dynamic, with characteristic changes during cancer progression. In a Chinese cohort, fecal samples from healthy individuals were dominated by Bacteroidetes and Firmicutes, the abundance of which decreased with progression along the polyp–adenoma–carcinoma sequence. In contrast, the abundance of Proteobacteria was noted to increase with colon cancer development [42]. Such shifts have even been seen in the relative abundance of individual bacterial taxa. In particular, Firmicutes, Actinobacteria, Lachnospiraceae, and the genus Desulfovibrio have been shown to be specific to early-stage CRC, while the genera Solobacterium, Peptostreptococcus, Corynebacterium, Parvimonas, Neisseria, Porphyromonas, Gemella, and the families Alcaligenaceae and Enterobacteriaceae appear to be associated with malignancy [37][43][44][45][37,43,44,45] (Figure 2).
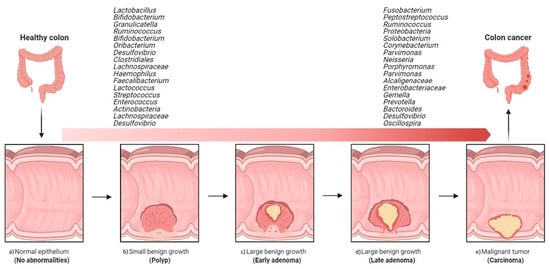
Figure 2. Overview of the implications of gut bacteria in the development and progression of colorectal cancer. Shift from healthy intestine to CRC intestine: (a) normal epithelium, (b) poly P (small benign growth), (c) early adenoma, (d) late adenoma, and (e) carcinoma. (The figure was created with Biorender.com).
3.1. Inflammation and CRC
Chronic inflammation is an established risk factor for CRC, as patients with inflammatory bowel diseases (IBD) consistently have a higher risk than the general population of developing CRC [46][47][46,47]. Correspondingly, an increase in pro-inflammatory species has been repeatedly reported in CRC patients (Figure 3). The most prevalent and most described bacterium in CRC fecal and mucosa-associated microbiota is Fusobacterium nucleatum [45], which, in murine models, increases the proliferation of CRC cells and colonic tumorigenesis by activating TLR4 signaling to NF-κB, thus promoting the infiltration of specific pro-inflammatory myeloid cell subsets into tumors [48][49][50][48,49,50]. Interestingly, a recent study found that more than 40% of CRC patients exhibited identical strains of F. nucleatum in both tumor and saliva samples, suggesting that F. nucleatum in CRC originates from the oral cavity [51]. In this, F. nucleatum does not appear to be alone; meta-analyses of geographically and technically diverse cohorts have identified several oral commensal and pathogenic bacteria that are significantly enriched in CRC samples, including members of the genera Fusobacterium, Porphyromonas, Parvimonas, Peptostreptococcus, Gemella, Prevotella, and Solobacterium. Taken together, these results reinforce the hypothesis of an oral–gut translocation route that is associated with inflammation and CRC [39][52][53][54][55][39,52,53,54,55]. Other examples of well-known pro-inflammatory species with links to CRC are colibactin-producing Escherichia coli, which enhances inflammation and the production of reactive oxygen species (ROS) in tumors in early-stage CRC [56], and enterotoxigenic Bactoroides fragilis, which mediates inflammation through the Th17 response and NF-κB activation, thus inducing myeloid cell-dependent distal colon tumorigenesis [57]. The increase in pro-inflammatory species observed in CRC correlates with a reduction in anti-inflammatory species belonging to the beneficial genera Ruminococcus, Bifidobacterium, Lachnospira, Oribacterium, Desulfovibrio, Clostridiales, and Lactobacillus [44][55][58][44,55,58]. Furthermore, the alterations observed in CRC with respect to microbiota composition may also translate into changes in metabolite concentrations. Specifically, a metabolomic study detected a direct association between significantly lower abundances of Clostridia and Lachnospiraceae in CRC and reduced quantities of the metabolites p-aminobenzoate and conjugated linoleic acid, which are known to exhibit anti-inflammatory and anti-cancerogenic properties [59].
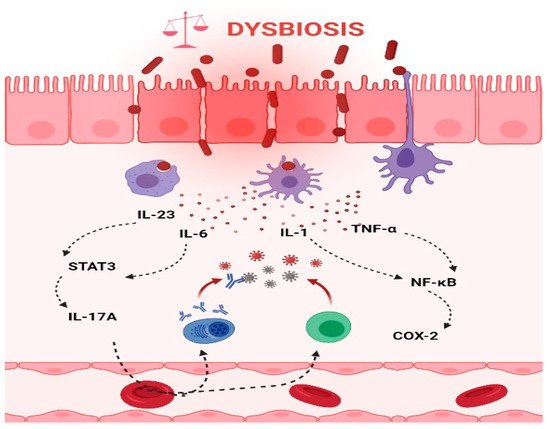
Figure 3. Intestinal inflammation caused by gut microbiota contributes to the onset of CRC. Dysbiotic bacteria can elicit immune imbalances and facilitate the translocation of gut microbiota and/or its metabolites due to a leaky gut to the tissues and systemic circulation. These events may lead to the stimulation of an inflammatory state and ultimately to the development of CRC. Thus, the production of IL-6 and IL-23, in turn, trigger the expression of IL-17A and contribute to the development of CRC through STAT3 activation. In addition, TNF-α and IL-1 promote pro-inflammatory and pro-tumorigenic activities of COX-2 that stimulate growth and angiogenesis and inhibit apoptosis in CRC. (The figure was created with Biorender.com).
3.2. DNA Damage and CRC
Certain members of the CRC-enriched microbiota are able to directly induce DNA damage to colonic epithelial cells. For example, some strains in the CRC-associated family Enterobacteriaceae produce ROS and colibactin, a toxin responsible for oncogenic mutations in host colonic epithelial cells [60][61][60,61]. The pivotal role of this toxin in carcinogenesis was confirmed by the finding that colibactin-producing E. coli promote tumorigenesis in ApcMin/+; IL10−/− mice in a colibactin-dependent manner [62]. A recent study identified a distinct mutational signature of genotoxic pks+ E. coli in human intestinal organoids, and this same signature was also detected in human CRC genomes. This suggests that the underlying mutational process may be the direct result of past exposure to bacteria that carried the colibactin-producing pks pathogenicity island [60]. However, E. coli is far from the only species with this capability. Molecular studies of F. nucleatum have discovered the virulence protein FadA and its involvement in the transformation of epithelial cells and the promotion of colon tumorigenesis [63]. A meta-analysis of CRC fecal metagenomes confirmed the significant enrichment in both the colibactin-producing gene cluster pks and the F. nucleatum adhesin fadA [53]. In vitro, enterotoxins produced by B. fragilis have been associated with DNA damage and genomic instability [64], while, in mice, Peptostreptococcus anaerobius, another CRC-enriched species, was shown to interact with TLR2/4 receptors on host cells to induce ROS production, increasing cholesterol biosynthesis and activating pro-oncogenic factors and CRC-promoting pathways [65]. CRC patients present increased amounts of sulphate-reducing bacteria belonging to genus Desulfovibrio [37][44][37,44], which explains the elevated levels in late-stage CRC of dissimilatory sulfate reductase subunit A, a gene responsible for the production of genotoxic hydrogen sulfide [37]. Multiple metabolomic analyses have reported increases in polyamines, such as putrescine and cadaverine, in CRC fecal samples compared to healthy controls [41][64][41,64]; in particular, the polyamine spermidine is known to promote colibactin-associated genotoxicity [66]. Finally, evidence from whole metagenomics analysis has linked the relative abundance of some members of the CRC microbiota with the methylation or demethylation of host genes, indicating that epigenome dysregulation may be another means by which CRC-associated dysbiosis promotes colon carcinogenesis [58]. For example, the B. fragilis toxin is able to induce epigenetic changes in vitro in HT-29 colon epithelial cells. The toxin alters the expression of specific genes, the accessibility of certain transcription factor binding sites, and the coordination between regions with different degrees of methylation, which, together, increases the risk of colon tumorigenesis [67].
3.3. Short-Chain Fatty Acids and CRC
Short-chain fatty acids (SCFAs) are the primary end-products of the fermentation of polysaccharides and nondigestible carbohydrates that remain available to the gut microbiota. Butyrate, acetate, and propionate are the most abundant SCFAs. Butyrate, in particular, has a remarkable array of colonic health-promoting and antineoplastic properties; along with being the preferred energy source for colonocytes, it maintains mucosal integrity, reduces pro-inflammatory cytokines, and induces apoptosis in CRC cell lines [68]. Compared to healthy controls, the fecal microbiota of patients with CRC and advanced colorectal adenoma demonstrates significant reductions in the abundance of butyrate-producing bacteria [55][69][55,69], and these reductions are dependent on CRC progression. Notably, the abundance of Oscillospira declines in the transition from advanced adenoma to stage 0 CRC, whereas levels of Haemophilus decrease in the transition from stage 0 to early-stage CRC [45]. A meta-analysis of fecal metagenomes confirmed a significant decrease in the carbohydrate-degradation genes responsible for SCFA production in CRC [53]. These changes in the microbiome and metagenome coincide with a decrease in butyrate concentration in CRC patients [41][69][41,69].
3.4. Bile Acid Metabolism and CRC
Primary bile acids are synthesized in the liver, conjugated to taurine or glycine, and released in the gut. Upon reaching the colon, bile acids are deconjugated by bile salt hydrolases of the gut microbiota and are subsequently transformed into dangerous secondary bile acids by 7α-dehydroxylating bacteria [70]. Alterations in this process have been associated with CRC. Metabolomic profiling confirmed the presence of elevated levels of secondary bile acids, including deoxycholic acid (DCA), in adenomas and/or intramucosal carcinomas [37][71][37,71]. In mice, DCA has been found to induce alterations in the gut microbiota that are accompanied by impairments in the intestinal barrier, low-grade inflammation, and colonic tumors [72][73][72,73]. DCA-induced dysbiosis is characterized by an increased abundance of pathogens and a decreased abundance of probiotics, and this shift in microbial community structure can be sufficient by itself to cause disease independent of DCA treatment [73]. Instead, the secondary ursodeoxycholic acid (UDCA), known for its anti-carcinogenic properties, is less abundant in CRC patients [69][74][69,74].
Generally speaking, a high-fat and low-fiber diet has long been known to represent a risk factor for CRC; specifically, this diet correlates with lower levels of colonic SCFAs and higher levels of colonic secondary bile acids and mucosal proliferative biomarkers of cancer risk [75]. Interestingly, directed dietary changes (switch from high-fat/low-fiber to low-fat/high-fiber diet and vice versa) resulted in remarkable reciprocal changes in mucosal biomarkers of cancer risk, with an increased saccharolytic fermentation and butyrogenesis and a suppressed secondary bile acid synthesis in the low-fat/high-fiber diet group [75].