Interleukin-1β (IL-1β) and type I interferons (IFNs) are major cytokines involved in autoinflammatory/autoimmune diseases. Separately, the overproduction of each of these cytokines is well described and constitutes the hallmark of inflammasomopathies and interferonopathies, respectively. While their interaction and the crosstalk between their downstream signaling pathways has been mostly investigated in the frame of infectious diseases, little information on their interconnection is still available in the context of autoinflammation promoted by sterile triggers.
- inflammation
- type I interferons
- interleukin-1β
- crosstalk
1. Introduction
Numerous reports have documented the roles of IL-1β and type I interferons (IFNs) in the defense mechanisms that are engaged upon bacterial (such as M. tuberculosis [1]) and viral [2] infections. Type I (and type III) IFNs exert powerful antiviral activities that have been extensively described [3][4], while those mediated by IL-1β are more scarcely defined [5]. Furthermore, the interplay of these cytokines and their downstream signaling pathways has also been largely explored during infectious diseases [6], COVID-19 being the most recent example [7].
These cytokines are produced following the activation of dedicated pattern-recognition receptors (PRRs) [8] in response to specific pathogens and the associated molecular patterns (PAMPs) that they express. Interestingly, the same PRRs (nucleotide-binding oligomerization domain-like receptors—NLRs, Toll-like receptors—TLR or AIM2-like receptors—ALRs) are also activated upon the detection of danger signals (DAMPs [9][10][11]) produced in sterile conditions. In this case, inflammation, instead of creating the appropriate conditions to clear off an invading pathogen, generates tissue damage and evolves towards detrimental endpoints for the host. First, this review will provide some examples of autoimmune/autoinflammatory diseases that are caused by the deregulated expression of type I IFNs and IL-1β. Indeed, these cytokines are major mediators of inflammation and can be incriminated in many cytokinopathies [12], which are diseases caused by alterations in a single gene affecting cytokines expression. Several examples of interferonopathies and inflammasomopathies will illustrate these cases. Additionally, type I IFNs and IL-1β perturbations can also result from interactions between many genes and the host environment. Lupus, a disease in which patients exhibit an “IFN signature” [13] (i.e., overexpression of a subset of IFN-stimulated genes) and Alzheimer’s disease, during which IL-1β is known to be overexpressed [14], will serve as examples for such complex (multigenic/multifactorial) diseases in which these cytokines are involved. Next, we will analyze several cases where reciprocal interactions between them have been observed, and the therapeutic perspectives that have been derived from these observations. Multiple sclerosis, a disease treated with IFN-β (among other therapeutic options) and which is also characterized by increased IL-1β expression, will be described. In parallel, gout and rheumatoid arthritis (RA) are joint inflammatory diseases in which reducing IL-1β overexpression can represent an efficient therapeutic opportunity. Interestingly, promoting type I IFNs expression recently appeared as an attractive way to dampen IL-1β production in animal models for gout and RA [15][16]. These examples in which type I IFNs and IL-1β exert a reciprocal control will reveal novel options to treat patients suffering from these inflammatory diseases, whose general features are given in Table 1. Finally, innovative cell culture methods designed to investigate and aimed at deciphering these interactions between cytokines at the molecular and cellular levels will be discussed in a prospective chapter.
Table 1. Type I IFNs- and IL-1β-mediated pathologies discussed in this review.
| Disease | Type | Genetic Defect | Cytokine Profile | Treatment |
|---|---|---|---|---|
| STING-associated vasculopathy with onset in infancy (SAVI) | interferonopathy | STING gain-of-function | exessive type I IFN secretion | corticosteroids, jakinhibs (clinical trials) |
| Systemic Lupus Erythematosus (SLE) | rheumatic autoimmune/autoinflammatory disease | multifactorial disease | IFN signature (overexpression of IFN-stimulated genes) | corticosteroids, Immunosuppressants (e.g., methotrexate), biologics (e.g antiB-cell mAb) |
| Familial Mediterranean Fever (FMF) | inflammasomopathy | mutations in MEFV (Mediterranean fever, also named PYRIN) | constitutive IL-1β secretion | colchicin, biologics (IL-1β receptor antagonist, anti IL-1β mAb) |
| Alzheimer’s disease (AD) | Neurodegenerative disease | multifactorial disease | excessive IL-1β, IL-6 and TNF secretion | Cholinesterase inhibitors, N-methyl D-aspartate (NMDA) antagonists, anti amyloid-β mAb (clinical trials) |
| Gout | rheumatic autoinflammatory disease | multifactorial disease | excessive IL-1β secretion | colchicin, biologics (IL-1β receptor antagonist, anti IL-1β mAb) |
| Rheumatoid Arthritis (RA) | rheumatic autoimmune/autoinflammatory disease | multifactorial disease | TNF overexpression IL-1β overexpression IFN signature (overexpression of IFN-stimulated genes) | corticosteroids, Immunosuppressants (e.g., methotrexate), biologics (e.g anti TNF mAb) |
| Multiple sclerosis (MS) | inflammatory, neurodegenerative disease | multifactorial disease | increased IFNγ, IL-12, IL-17 secretion/activation | IFN-β, biologics (e.g., antiB-cell mAb) |
2. Interplay between Type I IFNs and IL-1β in Inflammatory/Autoimmune Diseases
Whilst interferonopathies and inflammasomopathies may appear as very divergent or even antagonistic inflammatory diseases (although an overlap can be observed in some instances, as mentioned in the previous chapters), the pathogenesis of some inflammatory conditions clearly involves both type I IFNs and IL-1β. Multiple sclerosis (MS) belongs to this category, since IL-1β is strongly implicated in this inflammatory, neurodegenerative disease [17], and IFN-β is still a classical first-line therapy [18], although rituximab (an anti-CD20 monoclonal antibody designed to induce B cell ablation) was shown recently as a promising option [19]. Low STING-dependent type I IFNs expression in peripheral blood mononuclear cells (PBMC) isolated from MS patients [20] is in agreement with these observations.
The mechanism by which IFN-β exerts its anti-inflammatory actions has been partially elucidated [21]. It is now very clear that type I IFNs promote IFNAR-dependent IL-1Ra (encoding an antagonist of the IL-1β receptor) and IL-10 gene expressions. Furthermore, type I IFNs and IL-10 were recently shown to negatively regulate the activation of the NLRP3 inflammasome in a STAT3-dependent manner [22][23][24]. These data support the notion that IL-1β and type I IFNs exert antagonistic activities that have been experimentally tested in various inflammatory settings (collagen-induced arthritis, allotransplant rejection), whereby the beneficial administration of type I IFNs has been documented.
Reduced expression of NLRP3 was also shown to participate in the anti-inflammatory benefits of type I IFNs in MS [25][26]. This observation also likely accounts for the spectacular therapeutic potential of imiquimod, a TLR7 agonist and strong inducer of type I IFNs, which we observed in a mouse model of acute uratic inflammation [15]. Importantly, our work using this mouse model of gout as well as RA models [16] enabled us to develop a framework in which complex cellular interactions are required to account for the counter-regulatory effects mediated by type I IFNs on IL-1β [27]. Future work using elaborate cell culture systems will be necessary to decipher this cellular dialog, as discussed below. Surprisingly, the regulatory roles of IL-1β on type I IFNs and ISGs expression are more scarcely documented [28], and these experimental cell culture experiments would also be useful to explore this issue. In this regard, the recent observation that IL-1β promotes type I IFN and ISGs expression in bone marrow-derived dendritic cells (BMDC) appears of particular interest [6]. A schematic network of type I IFNs and IL-1β interactions is depicted in Figure 1.
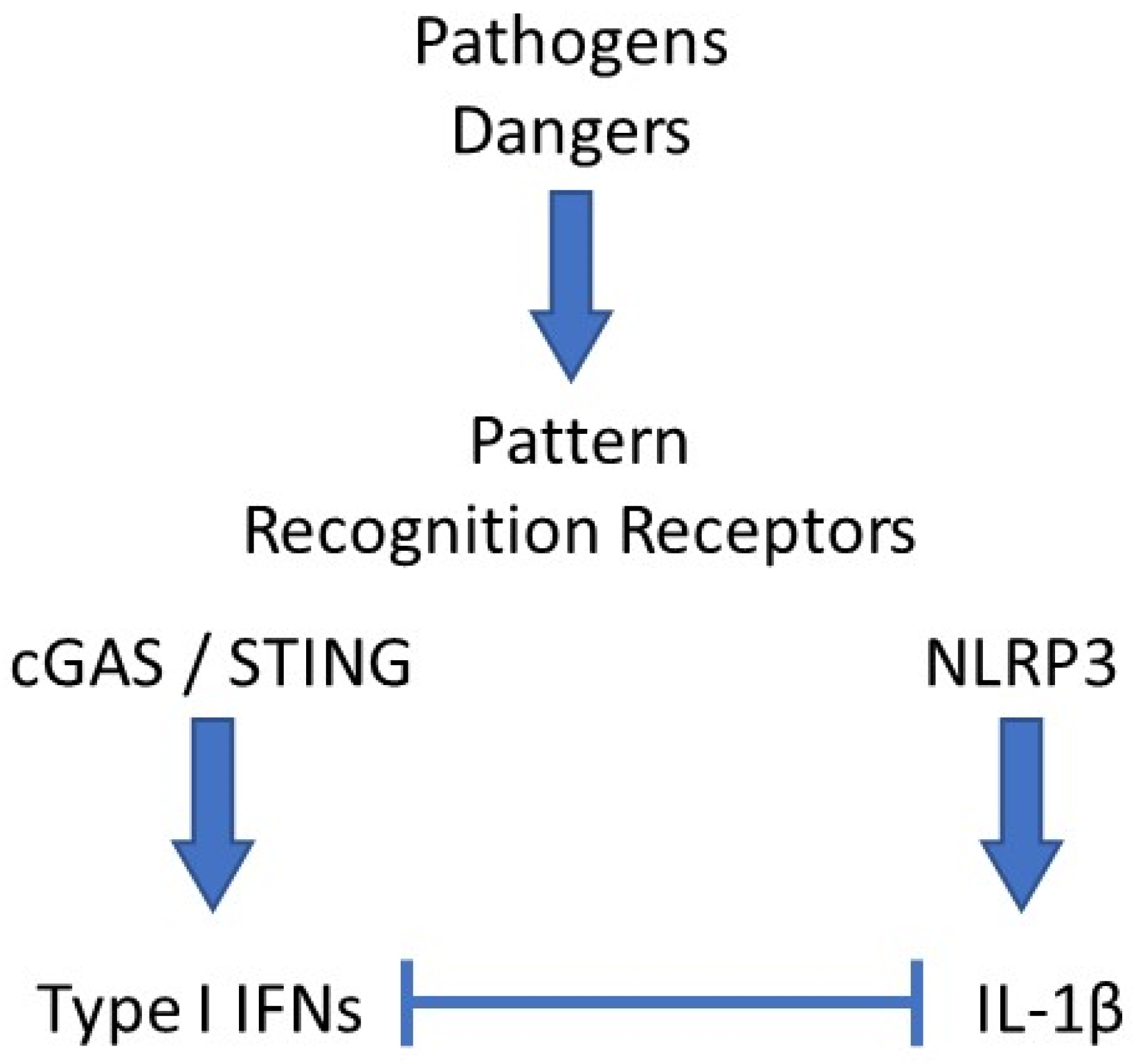
Figure 1. Schematic network of interactions between type I IFNs and IL-1β. Pathogens- or danger-associated molecular patterns (PAMPs, DAMPs) interact with their cognate pattern recognition receptor (PRRs). In the example shown here, DNA binding and activation of the cGAS/STING pathway leads to type I interferons (IFNs) secretion, while monosodium urate (MSU) crystals activate the NLRP3 inflammasome, which induces IL-1β release. In most cases, both cytokines exert antagonistic activities, mutually repressing their expression levels by various mechanisms.
References
- Ma, J.; Zhao, S.; Gao, X.; Wang, R.; Liu, J.; Zhou, X.; Zhou, Y. The Roles of Inflammasomes in Host Defense against Mycobacterium tuberculosis. Pathogens 2021, 10, 120.
- Schoggins, J.W. Recent advances in antiviral interferon-stimulated gene biology. F1000Research 2018, 7, 309.
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923.
- Li, S.-F.; Gong, M.-J.; Zhao, F.-R.; Shao, J.-J.; Xie, Y.-L.; Zhang, Y.-G.; Chang, H.-Y. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell. Physiol. Biochem. 2018, 51, 2377–2396.
- Orzalli, M.H.; Smith, A.; Jurado, K.A.; Iwasaki, A.; Garlick, J.A.; Kagan, J.C. An Antiviral Branch of the IL-1 Signaling Pathway Restricts Immune-Evasive Virus Replication. Mol. Cell 2018, 71, 825–840.
- Aarreberg, L.D.; Wilkins, C.; Ramos, H.J.; Green, R.; Davis, M.A.; Chow, K.; Gale, M. Interleukin-1beta Signaling in Dendritic Cells Induces Antiviral Interferon Responses. mBio 2018, 9, 2.
- Jamilloux, Y.; Henry, T.; Belot, A.; Viel, S.; Fauter, M.; El Jammal, T.; Walzer, T.; François, B.; Sève, P. Should we stimulate or suppress immune responses in COVID-19? Cytokine and anti-cytokine interventions. Autoimmun. Rev. 2020, 19, 102567.
- Kawai, T.; Akira, S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol. 2009, 21, 317–337.
- Land, W.G. The Role of Damage-Associated Molecular Patterns in Human Diseases: Part I—Promoting inflammation and immunity. Sultan Qaboos Univ. Med. J. 2015, 15, e9–e21.
- Land, W.G. The Role of Damage-Associated Molecular Patterns (DAMPs) in Human Diseases: Part II: DAMPs as diagnostics, prognostics and therapeutics in clinical medicine. Sultan Qaboos Univ. Med. J. 2015, 15, e157–e170.
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112.
- Moghaddas, F.; Masters, S.L. Monogenic autoinflammatory diseases: Cytokinopathies. Cytokine 2015, 74, 237–246.
- Szymczak, F.; Colli, M.L.; Mamula, M.J.; Evans-Molina, C.; Eizirik, D.L. Gene expression signatures of target tissues in type 1 diabetes, lupus erythematosus, multiple sclerosis, and rheumatoid arthritis. Sci. Adv. 2021, 7, eabd7600.
- Batista, A.F.; Rody, T.; Forny-Germano, L.; Cerdeiro, S.; Bellio, M.; Ferreira, S.T.; Munoz, D.P.; De Felice, F.G. Interleukin-1beta mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-beta oligomers. J. Neuroinflam. 2021, 18, 54.
- Mariotte, A.; De Cauwer, A.; Po, C.; Abou-Faycal, C.; Pichot, A.; Paul, N.; Aouadi, I.; Carapito, R.; Frisch, B.; Macquin, C.; et al. A mouse model of MSU-induced acute inflammation in vivo suggests imiquimod-dependent targeting of Il-1beta as relevant therapy for gout patients. Theranostics 2020, 10, 2158–2171.
- Nehmar, R.; Alsaleh, G.; Voisin, B.; Flacher, V.; Mariotte, A.; Saferding, V.; Puchner, A.; Niederreiter, B.; Vandamme, T.; Schabbauer, G.; et al. Therapeutic Modulation of Plasmacytoid Dendritic Cells in Experimental Arthritis. Arthritis Rheumatol. 2017, 69, 2124–2135.
- Musella, A.; Fresegna, D.; Rizzo, F.R.; Gentile, A.; De Vito, F.; Caioli, S.; Guadalupi, L.; Bruno, A.; Dolcetti, E.; Buttari, F.; et al. ’Prototypical’ proinflammatory cytokine (IL-1) in multiple sclerosis: Role in pathogenesis and therapeutic targeting. Expert Opin. Targets 2020, 24, 37–46.
- McGinley, P.M.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis: A Review. JAMA 2021, 325, 765–779.
- Chisari, C.G.; Sgarlata, E.; Arena, S.; Toscano, S.; Luca, M.; Patti, F. Rituximab for the treatment of multiple sclerosis: A review. J. Neurol. 2021, 1–25.
- Masanneck, L.; Eichler, S.; Vogelsang, A.; Korsen, M.; Wiendl, H.; Budde, T.; Meuth, S.G. The STING-IFN-beta-Dependent Axis Is Markedly Low in Patients with Relapsing-Remitting Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 9249.
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I Interferon Inhibits Interleukin-1 Production and Inflammasome Activation. Immunity 2011, 34, 213–223.
- Mayer-Barber, K.D.; Yan, B. Clash of the Cytokine Titans: Counter-regulation of interleukin-1 and type I interferon-mediated inflammatory responses. Cell. Mol. Immunol. 2017, 14, 22–35.
- Ludigs, K.; Parfenov, V.; Du Pasquier, R.A.; Guarda, G. Type I IFN-mediated regulation of IL-1 production in inflammatory disorders. Cell. Mol. Life Sci. 2012, 69, 3395–3418.
- Van Kempen, T.S.; Wenink, M.H.; Leijten, E.F.; Radstake, T.R.; Boes, M. Perception of self: Distinguishing autoimmunity from autoinflammation. Nat. Rev. Rheumatol. 2015, 11, 483–492.
- Malhotra, S.; Costa, C.; Eixarch, H.; Keller, C.W.; Amman, L.; Martínez-Banaclocha, H.; Midaglia, L.; Sarró, E.; Machín-Díaz, I.; Villar, L.M.; et al. NLRP3 inflammasome as prognostic factor and therapeutic target in primary progressive multiple sclerosis patients. Brain 2020, 143, 1414–1430.
- Piancone, F.; Saresella, M.; Marventano, I.; La Rosa, F.; Santangelo, M.A.; Caputo, D.; Mendozzi, L.; Rovaris, M.; Clerici, M. Monosodium Urate Crystals Activate the Inflammasome in Primary Progressive Multiple Sclerosis. Front. Immunol. 2018, 9, 983.
- Nehmar, R.; Mariotte, A.; De Cauwer, A.; Sibilia, J.; Bahram, S.; Georgel, P. Therapeutic Perspectives for Interferons and Plasmacytoid Dendritic Cells in Rheumatoid Arthritis. Trends Mol. Med. 2018, 24, 338–347.
- Kohase, M.; Zhang, Y.; Lin, J.X.; Yamazaki, S.; Sehgal, P.B.; Vilček, J. Interleukin-1 can inhibit interferon-beta synthesis and its antiviral action: Comparison with tumor necrosis factor. J. Interferon. Res. 1988, 8, 559–570.