Symptomatic treatments are available for Parkinson’s disease and Alzheimer’s disease. An unmet need is cure or disease modification. This review discusses possible reasons for negative clinical study outcomes on disease modification following promising positive findings from experimental research. It scrutinizes current research paradigms for disease modification with antibodies against pathological protein enrichment, such as α-synuclein, amyloid or tau, based on post mortem findings. Instead a more uniform regenerative and reparative therapeutic approach for chronic neurodegenerative disease entities is proposed with stimulation of an endogenously existing repair system, which acts independent of specific disease mechanisms. The repulsive guidance molecule A pathway is involved in the regulation of peripheral and central neuronal restoration. Therapeutic antagonism of repulsive guidance molecule A reverses neurodegeneration according to experimental outcomes in numerous disease models in rodents and monkeys. Antibodies against repulsive guidance molecule A exist. First clinical studies in neurological conditions with an acute onset are under way. Future clinical trials with these antibodies should initially focus on well characterized uniform cohorts of patients. The efficiency of repulsive guidance molecule A antagonism and associated stimulation of neurogenesis should be demonstrated with objective assessment tools to counteract dilution of therapeutic effects by subjectivity and heterogeneity of chronic disease entities. Such a research concept will hopefully enhance clinical test strategies and improve the future therapeutic armamentarium for chronic neurodegeneration
Symptomatic treatments are available for Parkinson’s disease and Alzheimer’s disease. An unmet need is cure or disease modification. This review discusses possible reasons for negative clinical study outcomes on disease modification following promising positive findings from experimental research. It scrutinizes current research paradigms for disease modification with antibodies against pathological protein enrichment, such as -synuclein, amyloid or tau, based on post mortem findings. Instead a more uniform regenerative and reparative therapeutic approach for chronic neurodegenerative disease entities is proposed with stimulation of an endogenously existing repair system, which acts independent of specific disease mechanisms. The repulsive guidance molecule A pathway is involved in the regulation of peripheral and central neuronal restoration. Therapeutic antagonism of repulsive guidance molecule A reverses neurodegeneration according to experimental outcomes in numerous disease models in rodents and monkeys. Antibodies against repulsive guidance molecule A exist. First clinical studies in neurological conditions with an acute onset are under way. Future clinical trials with these antibodies should initially focus on well characterized uniform cohorts of patients. The efficiency of repulsive guidance molecule A antagonism and associated stimulation of neurogenesis should be demonstrated with objective assessment tools to counteract dilution of therapeutic effects by subjectivity and heterogeneity of chronic disease entities. Such a research concept will hopefully enhance clinical test strategies and improve the future therapeutic armamentarium for chronic neurodegeneration- neurodegeneration
- repulsive guidance molecule A
- neuroprotection
- repair
- oxidative stress
- apoptosis
- neurogenesis
1. Introduction
1. Introduction One of the main causes for disability in humans worldwide is onset of neurological disorders, such as stroke and chronic progressive neurodegenerative brain diseases (PND). The most prevalent PNDs are the idiopathic and genetic Parkinson’s disease entity (PD) and the complex of various dementia syndromes, mainly consisting of Alzheimer’s disease (AD), frontotemporal dementia (FTD), mixed dementia (MD) and vascular dementia (VD) (1-3). They are characterized by a common pathophyiologic mechanism, which is aberrant protein aggregation. Well known neuropathological features are β-amyloid and tau-protein enrichment in AD and accumulation of misfolded -synuclein in PD (4;5). Incidence of these PNDs will further rise. As an example, estimates of PD prevalence showed a 2.4 fold rise in the last 30 years. Main reasons are an earlier diagnosis associated with better treatment quality and a general rise of human life ex-pectancy (6). Increased exposure to endogenous and exogenous toxins contributes to a slowly evolving neurodegeneration in the peripheral and central nervous system and accelerates the overall ageing process in a pathological PND related manner. Typical risk factors are pesticides or herbicides, paraquat, rotenone, various metals (i.e. iron, manganese, lead), gaseous compounds (such as carbon monoxide) and even viruses (7;8). The rising number of dementia- and PD patients will increase the financial burden for health care systems worldwide. To date, it is far from clear, whether the current SARS-CoV-2 outbreak may cause PND like syndromes in the long run, similar to the observed symptomatic PD forms following the 1918 H5N1 influenza pandemic (9).One of the main causes for disability in humans worldwide is onset of neurological disorders, such as stroke and chronic progressive neurodegenerative brain diseases (PND). The most prevalent PNDs are the idiopathic and genetic Parkinson’s disease entity (PD) and the complex of various dementia syndromes, mainly consisting of Alzheimer’s disease (AD), frontotemporal dementia (FTD), mixed dementia (MD) and vascular dementia (VD) [1][2][3]. They are characterized by a common pathophyiologic mechanism, which is aberrant protein aggregation. Well known neuropathological features are β-amyloid and tau-protein enrichment in AD and accumulation of misfolded a-synuclein in PD [4][5]. Incidence of these PNDs will further rise. As an example, estimates of PD prevalence showed a 2.4 fold rise in the last 30 years. Main reasons are an earlier diagnosis associated with better treatment quality and a general rise of human life expectancy [6]. Increased exposure to endogenous and exogenous toxins contributes to a slowly evolving neurodegeneration in the peripheral and central nervous system and accelerates the overall ageing process in a pathological PND related manner. Typical risk factors are pesticides or herbicides, paraquat, rotenone, various metals (i.e. iron, manganese, lead), gaseous compounds (such as carbon monoxide) and even viruses [7][8]. The rising number of dementia- and PD patients will increase the financial burden for health care systems worldwide. To date, it is far from clear, whether the current SARS-CoV-2 outbreak may cause PND like syndromes in the long run, similar to the observed symptomatic PD forms following the 1918 H5N1 influenza pandemic [9].
1.2. The current situation and unmet needs1.1. The current situation and unmet needs
Considerable research activities in the past 60 years have focused on symptomatic therapies for alleviation of PD. A success story was the introduction of the dopamine substitution concept. It alleviates motor and to a considerable extent associated non motor symptoms in PD since the 1960s (10;11). At that time Levodopa (L-dopa) was initially applied in an appropriate dose. The introduction of L-dopa therapy was based on findings, that high dopamine levels exist in the basal ganglia and that the dopamine precursor L-dopa counteracts reserpine induced dopamine decrease and associated impaired motor behavior (for review: (12)). Similar to PD, non motor symptoms also gained more and more interest in recent years in dementia. A considerable overlap exists between mechanisms of disease progression between PD and dementia syndromes. Thus the former focus on the dopamine deficiency in PD, respectively the acetylcholine deficit in AD is superseded by a more wide spread view. It also considers the individual different decline of other neurotransmitter systems, like serotonine (5-HT) or norepinephrine (13;14) Generally, particularly AD and PD are related to each other, i.e. by signs of microglial activation and neuroinflammation, and even in terms of neuropathological abnormalities (15-17). Similar therapeutic approaches are also employed. As an example, acetylcholine esterase inhibiting compounds and glutamate neurotransmission reducing drugs improve cognitive abilities not only in AD, MD and VD but also in PD plus dementia syndromes (18-20).Considerable research activities in the past 60 years have focused on symptomatic therapies for alleviation of PD. A success story was the introduction of the dopamine substitution concept. It alleviates motor and to a considerable extent associated non motor symptoms in PD since the 1960s [10][11]. At that time Levodopa (L-dopa) was initially applied in an appropriate dose. The introduction of L-dopa therapy was based on findings, that high dopamine levels exist in the basal ganglia and that the dopamine precursor L-dopa counteracts reserpine induced dopamine decrease and associated impaired motor behavior (for review: [12]). Similar to PD, non motor symptoms also gained more and more interest in recent years in dementia. A considerable overlap exists between mechanisms of disease progression between PD and dementia syndromes. Thus the former focus on the dopamine deficiency in PD, respectively the acetylcholine deficit in AD is superseded by a more wide spread view. It also considers the individual different decline of other neurotransmitter systems, like serotonine (5-HT) or norepinephrine [13][14] Generally, particularly AD and PD are related to each other, i.e. by signs of microglial activation and neuroinflammation, and even in terms of neuropathological abnormalities [15][16][17]. Similar therapeutic approaches are also employed. As an example, acetylcholine esterase inhibiting compounds and glutamate neurotransmission reducing drugs improve cognitive abilities not only in AD, MD and VD but also in PD plus dementia syndromes [18][19][20].
2. Pitfalls of translational concepts in clinical research2. Pitfalls of translational concepts in clinical research
To date, extensive experimental and neuropathological research provided distinct and better insights and understanding of chronic neuronal and associated glial cell death. The predominant responsible and final mechanism cascades are well identified and described in detail (21). Based on these findings, i.e. antiapoptotic, neuroprotective or oxidative stress reducing compounds, were successfully tested in experimental chronic neurodegenerative and inflammatory disease models (4;22-24) However, translation into positive clinical study results has failed so far, as trials on cure or disease modification in PNDs were more or less negative. Even transplantation of neurons or administration of neuronal growth factors was negative (as examples: (25;26)). Stimulation of growth factor synthesis, gene modification and stem cell applications are still discussed as promising tools (27-35). The unmet need for disease modification, respectively repair regeneration for central nervous system disorders, still exists. The “Neuroplasticity” concept has been suggested to be responsible for the compensation of deleterious metabolic processes and the delayed occurrence of symptoms (36). Another critical issue is the therapeutic mode of action, which is utilized for disease modification or cure. As an example, antibodies against pathological misfolded proteins were developed based on neuropathological findings. Enrichment of these altered proteins, i.e. in Lewy bodies (LB) or plaques, are looked upon as the main responsible and important, pathological phenomenon in chronic neurodegenerative brain disorders, such as AD or PD (37). Failures within physiologic activities of protein metabolism may cause protein degradation and misfolding. However it is far from clear, whether these abnormalities represent a specific process, which is responsible for disease onset and progression (38). This pathologic protein accumulation may also be the result of an unspecific side reaction of the metabolic cascade during chronic neurodegenerative processes. It may hypothetically only represent well wrapped protein garbage as consequence of physiologic defence mechanisms (38). The extent of compensatory capacity, the triggering causes and the moment of initiation of these misfolded protein enrichments during the disease process are not known in detail. However there is consensus, that an essential clinical precondition for disease modifying therapeutic concepts is an early diagnosis, when the disease caused damage is low. To date, PD and Alzheimer’s disease (AD) are mostly diagnosed relatively late in the disease process due to the compensatory “neuro-plasticity” phenomenon in the brain. A treatment allocation following earlier prodromal diagnostic screening will also probably reduce the current abundant missing motivation of PND-at-risk individuals for a testing procedure (39).To date, extensive experimental and neuropathological research provided distinct and better insights and understanding of chronic neuronal and associated glial cell death. The predominant responsible and final mechanism cascades are well identified and described in detail [21]. Based on these findings, i.e. antiapoptotic, neuroprotective or oxidative stress reducing compounds, were successfully tested in experimental chronic neurodegenerative and inflammatory disease models [4][22][23][24] (Figure 1).
3. Conclusion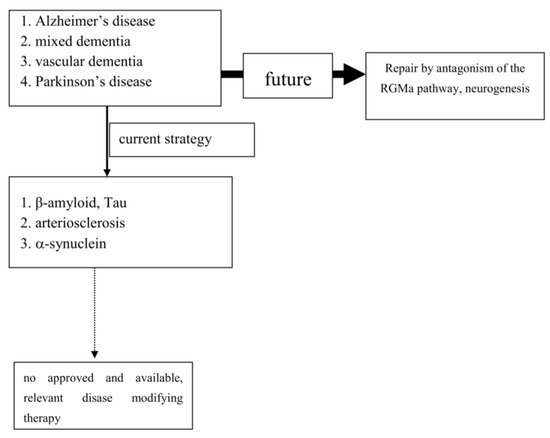
However, translation into positive clinical study results has failed so far, as trials on cure or disease modification in PNDs were more or less negative. Even transplantation of neurons or administration of neuronal growth factors was negative (as examples: [25][26]). Stimulation of growth factor synthesis, gene modification and stem cell applications are still discussed as promising tools [27][28][29][30][31][32][33][34][35]. The unmet need for disease modification, respectively repair regeneration for central nervous system disorders, still exists. The “Neuroplasticity” concept has been suggested to be responsible for the compensation of deleterious metabolic processes and the delayed occurrence of symptoms [36]. Another critical issue is the therapeutic mode of action, which is utilized for disease modification or cure. As an example, antibodies against pathological misfolded proteins were developed based on neuropathological findings. Enrichment of these altered proteins, i.e. in Lewy bodies (LB) or plaques, are looked upon as the main responsible and important, pathological phenomenon in chronic neurodegenerative brain disorders, such as AD or PD [37]. Failures within physiologic activities of protein metabolism may cause protein degradation and misfolding. However it is far from clear, whether these abnormalities represent a specific process, which is responsible for disease onset and progression [38]. This pathologic protein accumulation may also be the result of an unspecific side reaction of the metabolic cascade during chronic neurodegenerative processes. It may hypothetically only represent well wrapped protein garbage as consequence of physiologic defence mechanisms [38]. The extent of compensatory capacity, the triggering causes and the moment of initiation of these misfolded protein enrichments during the disease process are not known in detail. However there is consensus, that an essential clinical precondition for disease modifying therapeutic concepts is an early diagnosis, when the disease caused damage is low. To date, PD and Alzheimer’s disease (AD) are mostly diagnosed relatively late in the disease process due to the compensatory “neuroplasticity” phenomenon in the brain. A treatment allocation following earlier prodromal diagnostic screening will also probably reduce the current abundant missing motivation of PND-at-risk individuals for a testing procedure [39].
3. Conclusion
Cure or modification of progression in dementia, particularly AD, and PD is an important unmet need. Clinical trials, which aimed to translate promising experimental research outcomes into relevant positive results, were negative. No therapy has yet been approved despite well identified final main neuronal and related glial cell death mechanisms mostly on the cellular level. Multifactorial origins, heterogeneity of clinical symptoms, variability of each other complementing disease mechanisms and progression are the mostly likely reasons.
4. Outlook
Experimental research convincingly described a number of cellular pathways leading to chronic neuronal degeneration and death in PNDs. Examples are mitochondriopathy, dysfunction of the ubiquitin/proteasome system, oxidative and nitrosative stress, dysregulation of heat shock response, altered iron metabolism and vesicular transport systems, apoptosis, necrosis, autophagy, microglial activation combined with neuroinflammation [16][40][41]. Therapeutic concepts were and are developed based on findings, such as that the pro-inflammatory TNF-alpha cytokine is able to modify neuronal plasticity, maturation, and function of human cholinergic neurons also by epigenetic mechanisms [42]. All of these disease related alterations and their possible therapies will consequently change neurotransmission pathways [43]. However to date, preventive or PND progression delaying therapeutic strategies failed following translation into clinical study programs (as an example: [44]). Even the current clinical testing of specific antibodies against certain proteins, which accumulate in the various sporadic PNDs subtypes, may probably fail as shown in AD. This may suggest that the enrichment of these proteins is not specific. Their accumulation overlaps between various clinical PND pictures. This protein enrichment in LB may hypothetically only represent a defence mechanisms against the disease process itself, but do not cause it (Table 1; [45]). To date all clinical studies, which aimed to demonstrate neuroprotection or disease modification, i.e. in PD and AD, showed that research on a specific pathological disease mechanism does not lead to an essential therapeutic innovation in terms of disease course modification (Figure 1). Therefore, the underlying research method is worth to be considered for a modification. As an alternative to this misconception, one may consider the stimulation of an existing repair system in the peripheral and central nervous system as a more promising research paradigm [46][47][48][49][50][51]. Therapeutic strategies, which antagonize the repulsive guidance molecule A (RGMa) pathway, are worth for further development in clinical trials. A RGMa increase in the substantia nigra was found by in situ hybridization and immunohistochemistry in neuromelanin-positive neurons in post-mortem tissue of treated PD patients [47]. It may also be related to L-dopa administration and associated oxidative stress generation to a certain extent [52][53]. Extracellular RGMa inhibits axon regeneration and therefore may accelerate demise of neurons [54][55][56]. However targeting the RGMa pathway with antibodies or neutralisation, respectively antagonism of the neogenin receptor activity, may start regeneration not only in acute, but also in chronic inflammatory and neurodegenerative disorders [36][57][58][59][60] (Figure 1). It is well known, that considerable metabolic similarities exist both in the peripheral and central nervous system. Therefore, it is hypothesized that other syndromes than PD and AD, may also respond to this approach [49][51][55][61][62]. It may restore neuronal function in the long term as a general concept for repair and may weaken efficiency of toxin exposure [36][47][48][57][58][63][64][65]. Well designed clinical long term trials with RGMa antagonizing approaches are urgently needed in multiple sclerosis, PD, dementia syndromes, stroke, or spinal cord injury. Neuropathies (NP), diabetic retinopathy, Guillian Barre syndrome and amyotrophic lateral sclerosis are particularly suitable disorders. They allow testing of this approach in rather homogenous, well defined study cohorts with objective assessment tools, such as visual function and visual evoked potentials in retinopathy, or sensory or motor nerve conduction assessment in NP. RGMa antagonism may probably also help to counteract heterogeneous neurological deficits as consequence from severe viral infections, including SARS-CoV-2. Currently two different neutralizing RGMa antibodies (ABT-555; MT-3921) are in phase 2 clinical trials in spinal cord injury. In addition ABT-555 is in phase 2 clinical trials in progressive and relapse-remitting multiple sclerosis and in ischemic stroke. A positive outcome of these clinical trials will support this strategy for regeneration and repair in the damaged human nervous system. A further important pathological mechanism-of-action in PNDs is the potential inhibition of neurogenesis by the RGMa-neogenin pathway also in PD and dementia syndromes [46][49][51][66]. Neurogenesis also occurs in the adult human brain, i.e. in the dentate gyrus or the subventricular zone. RGMa blocks neurogenesis in these areas [55][67]. As shown in the hippocampal dentate gyrus, blocking of RGMa promoted formation of new neurons [46]. Targeting RGMa by antibodies may promote neurogenesis in the adult human brain of PND patients. An increased neurogenesis may also improve motor symptoms in PD or cognitive deficits in dementia. This is an alternative to cell replacement and stem cell concepts. Both have a focus on specific cell types only in contrast to the potential of RGMa antagonism in chronic neurodegeneration [68][28][69][70][71][72][73].